


HSV Replication: Triggering and Repressing STING Functionality


Abstract
:1. Introduction
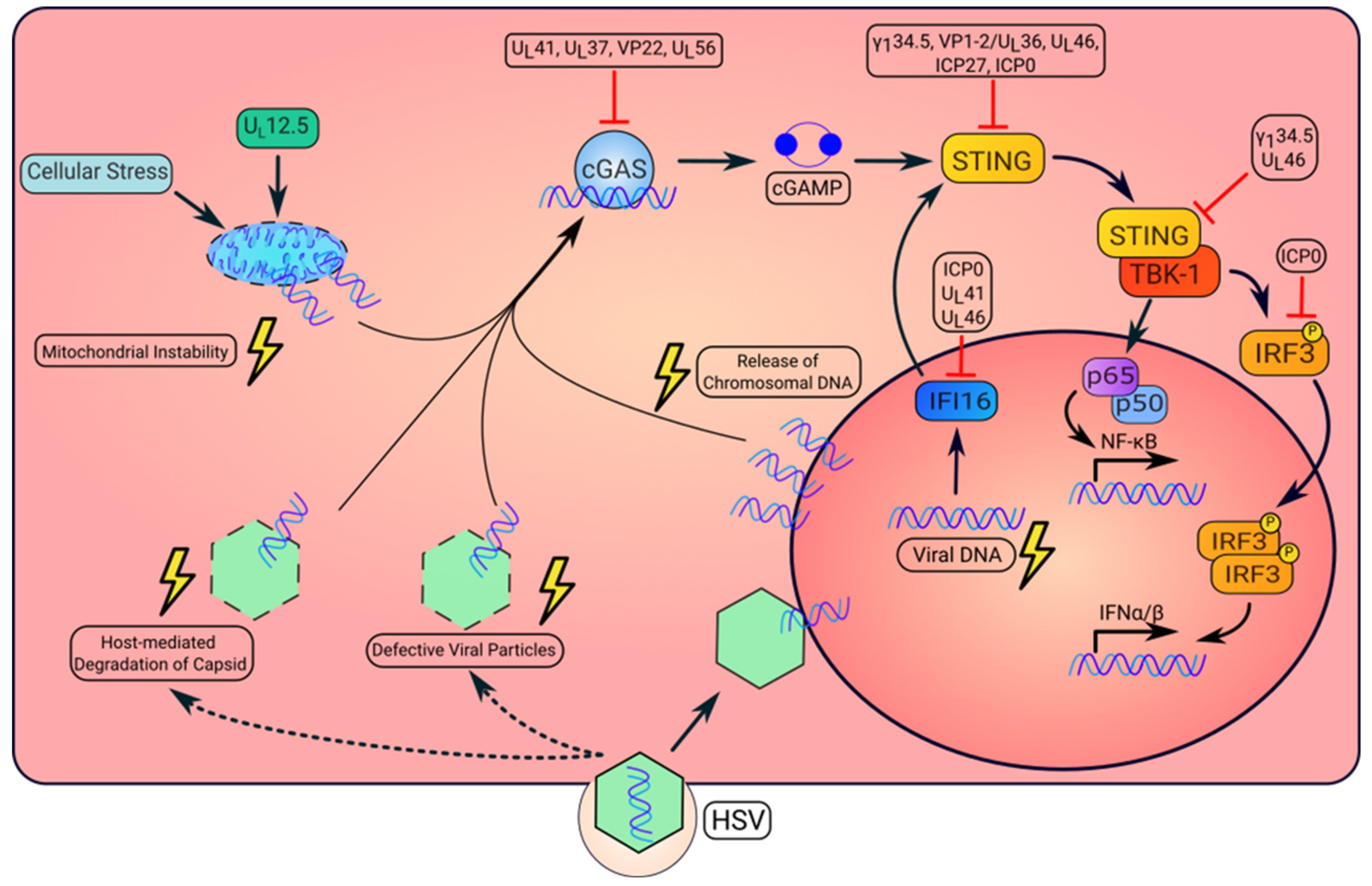
2. Induction of STING Activity by HSV
2.1. Regulation of STING Activation by DNA Sensors
2.2. Mechanisms of HSV Sensing
2.2.1. Mitochondrial DNA
2.2.2. Nuclear DNA
2.2.3. Viral DNA
3. HSV Interference of the STING Pathway
3.1. ICP0
3.2. ICP27
3.3. UL36/VP1-2
3.4. UL37
3.5. UL41
3.6. UL46
3.7. VP22 (UL49)
3.8. UL56
3.9. γ134.5

{kind=link}
| Viral Protein | Target Protein | Mechanism of Action | Reference |
|---|---|---|---|
| ICP0 | IFI16 | Interacts with and mediates IFI16 degradation | [55] |
| DNA-PK | Targets DNA-PK for proteasomal degradation | [61] | |
| ICP27 | STING | Interacts with the STING-TBK1-IRF3 complex | [107] |
| VP1-2/UL36USP | STING | Removes the K63-like polyubiquitin from STING | [28] |
| UL37 | cGAS | Deamidizes cGAS causing an impairment in cGAS ability to produce cGAMP | [113] |
| UL41 | cGAS | Targets cGAS mRNA for degradation | [114] |
| IFI16 | Targets IFI16 mRNA for degradation | [55] | |
| UL46 | TBK1 | Reduces dimerization of TBK1 impairing interaction with IRF3 | [119] |
| STING and IFI16 | Presence of UL46 causes elimination of STING and IFI16 | [122] | |
| VP22 | cGAS | Inhibits enzymatic activity of cGAS Forms a liquid condensation with DNA disrupting cGAS activity | [126] [127] |
| UL56 | cGAS | Interacts with cGAS to inhibit binding to viral DNA | [130] |
| γ134.5 | STING | Interacts with and blocks STING translocation from ER to the Golgi | [30] |
4. STING in HSV Replication and Pathogenesis
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Lafferty, W.E.; Downey, L.; Celum, C.; Wald, A. Herpes simplex virus type 1 as a cause of genital herpes: Impact on surveillance and prevention. J. Infect. Dis. 2000, 181, 1454–1457. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Warren, T.; Wald, A. Genital herpes. Lancet 2007, 370, 2127–2137. [Google Scholar] [CrossRef]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.-H.; Wang, X.; Wang, L.; Siedow, J.N.; Zhang, W.; Pei, Z.-M. Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res. 2014, 42, 8243–8257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranzusch, P.J.; Wilson, S.C.; Lee, A.S.; Berger, J.M.; Doudna, J.A.; Vance, R.E. Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2’,3’ cGAMP Signaling. Mol. Cell 2015, 59, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.R.; Wilson, S.C.; Vance, R.E. Evolutionary Origins of cGAS-STING Signaling. Trends Immunol. 2017, 38, 733–743. [Google Scholar] [CrossRef]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Liu, X.-Y.; Du, X.-X.; Jiang, Z.-F.; Su, X.-D. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol. 2012, 19, 728–730. [Google Scholar] [CrossRef]
- Yin, Q.; Tian, Y.; Kabaleeswaran, V.; Jiang, X.; Tu, D.; Eck Michael, J.; Chen, Z.J.; Wu, H. Cyclic di-GMP Sensing via the Innate Immune Signaling Protein STING. Mol. Cell 2012, 46, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Mukai, K.; Takaya, E.; Shindo, R. STING Operation at the ER/Golgi Interface. Front. Immunol. 2021, 12, 646304. [Google Scholar] [CrossRef]
- Sun, M.-S.; Zhang, J.; Jiang, L.-Q.; Pan, Y.-X.; Tan, J.-Y.; Yu, F.; Guo, L.; Yin, L.; Shen, C.; Shu, H.-B.; et al. TMED2 Potentiates Cellular IFN Responses to DNA Viruses by Reinforcing MITA Dimerization and Facilitating Its Trafficking. Cell Rep. 2018, 25, 3086–3098.e3083. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Xiong, M.-G.; Xu, Z.-S.; Luo, W.-W.; Wang, S.-Y.; Wang, Y.-Y. YIPF5 Is Essential for Innate Immunity to DNA Virus and Facilitates COPII-Dependent STING Trafficking. J. Immunol. 2019, 203, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Brandizzi, F.; Barlowe, C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 2013, 14, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.-W.; Li, S.; Li, C.; Lian, H.; Yang, Q.; Zhong, B.; Shu, H.-B. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 2016, 17, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef] [Green Version]
- Bodda, C.; Reinert, L.S.; Fruhwürth, S.; Richardo, T.; Sun, C.; Zhang, B.-c.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergström, P.; et al. HSV1 VP1–2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217, e20191422. [Google Scholar] [CrossRef]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding Kinase 1-mediated Signaling by the γ134.5 Protein of Herpes Simplex Virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ(1)34.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Smale, S.T. Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddeo, B.; Luo, T.R.; Zhang, W.; Roizman, B. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12408–12413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venuti, A.; Musarra-Pizzo, M.; Pennisi, R.; Tankov, S.; Medici, M.A.; Mastino, A.; Rebane, A.; Sciortino, M.T. HSV-1\EGFP stimulates miR-146a expression in a NF-κB-dependent manner in monocytic THP-1 cells. Sci. Rep. 2019, 9, 5157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e745. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, H.; Wang, C.; Li, Y.; Tian, H.; Siraj, S.; Sehgal, S.A.; Wang, X.; Wang, J.; Shang, Y.; et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e1118. [Google Scholar] [CrossRef]
- Yamashiro, L.H.; Wilson, S.C.; Morrison, H.M.; Karalis, V.; Chung, J.J.; Chen, K.J.; Bateup, H.S.; Szpara, M.L.; Lee, A.Y.; Cox, J.S.; et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat. Commun. 2020, 11, 3382. [Google Scholar] [CrossRef]
- Wu, Y.; Song, K.; Hao, W.; Li, J.; Wang, L.; Li, S. Nuclear soluble cGAS senses double-stranded DNA virus infection. Commun. Biol. 2022, 5, 433. [Google Scholar] [CrossRef]
- Zhang, X.; Bai, X.-c.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhu, Y.; Liu, Z.J.; Ouyang, S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell 2017, 8, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.S.; Vidhyasagar, V.; Yang, S.; Arna, A.B.; Yadav, M.; Aggarwal, A.; Aguilera, A.N.; Shinriki, S.; Bhanumathy, K.K.; Pandey, K.; et al. DDX41 is required for cGAS-STING activation against DNA virus infection. Cell Rep. 2022, 39, 110856. [Google Scholar] [CrossRef]
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 1999, 73, 650–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M., Jr.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Goulet, M.L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Menotti, L.; Avitabile, E.; Gianni, T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr Opin Virol 2012, 2, 28–36. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, D.S. Effect of herpes simplex virus type 2 infection on mitochondrial gene expression. J. Gen. Virol. 1988, 69 Pt 6, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lund, K.; Ziola, B. Synthesis of mitochondrial macromolecules in herpes simplex type 1 virus infected Vero cells. Biochem. Cell Biol. 1986, 64, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, T.; Lehman, I.R. Release of RNA polymerase from vero cell mitochondria after herpes simplex virus type 1 infection. J. Virol. 1990, 64, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, G.; Luo, R.; Lei, J.; Song, Y.; Wang, B.; Ma, L.; Liao, Z.; Ke, W.; Liu, H.; et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp. Mol. Med. 2022, 54, 129–142. [Google Scholar] [CrossRef]
- Berry, N.; Suspène, R.; Caval, V.; Khalfi, P.; Beauclair, G.; Rigaud, S.; Blanc, H.; Vignuzzi, M.; Wain-Hobson, S.; Vartanian, J.-P. Herpes Simplex Virus Type 1 Infection Disturbs the Mitochondrial Network, Leading to Type I Interferon Production through the RNA Polymerase III/RIG-I Pathway. mBio 2021, 12, e02557-21. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Monier, K.; Armas, J.C.; Etteldorf, S.; Ghazal, P.; Sullivan, K.F. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Holley, M.; Colgrove, R.C.; Nalepa, G.; Harper, J.W.; Knipe, D.M. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 2005, 79, 12840–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siew, V.-K.; Duh, C.-Y.; Wang, S.-K. Human cytomegalovirus UL76 induces chromosome aberrations. J. Biomed. Sci. 2009, 16, 107. [Google Scholar] [CrossRef]
- Evilevitch, A.; Hohlbauch, S.V. Intranuclear HSV-1 DNA ejection induces major mechanical transformations suggesting mechanoprotection of nucleus integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2114121119. [Google Scholar] [CrossRef]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; García Arcos, J.M.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e822. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Nava, M.M.; Wickström, S.A. Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci. 2017, 130, jcs.202192. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- De Chiara, G.; Racaniello, M.; Mollinari, C.; Marcocci, M.E.; Aversa, G.; Cardinale, A.; Giovanetti, A.; Garaci, E.; Palamara, A.T.; Merlo, D. Herpes Simplex Virus-Type1 (HSV-1) Impairs DNA Repair in Cortical Neurons. Front. Aging Neurosci. 2016, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Luecke, S.; Bodda, C.; Jønsson, K.L.; Cai, Y.; Zhang, B.-C.; Jensen, S.B.; Nordentoft, I.; Jensen, J.M.; Jakobsen, M.R.; et al. Cellular Requirements for Sensing and Elimination of Incoming HSV-1 DNA and Capsids. J. Interf. Cytokine Res. 2019, 39, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal Degradation of Herpes Simplex Virus Capsids in Macrophages Releases DNA to the Cytosol for Recognition by DNA Sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.C.; Dybas, J.M.; Hughes, J.; Weitzman, M.D.; Boutell, C. The HSV-1 ubiquitin ligase ICP0: Modifying the cellular proteome to promote infection. Virus Res. 2020, 285, 198015. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000, 74, 2052–2056. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The Viral Ubiquitin Ligase ICP0 Is neither Sufficient nor Necessary for Degradation of the Cellular DNA Sensor IFI16 during Herpes Simplex Virus 1 Infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [Green Version]
- Full, F.; Ensser, A. Early Nuclear Events after Herpesviral Infection. JCM 2019, 8, 1408. [Google Scholar] [CrossRef] [Green Version]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular Localization of the Herpes Simplex Virus ICP0 Protein Dictates Its Ability to Block IRF3-Mediated Innate Immune Responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef]
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-β induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. USA 2014, 111, E611–E617. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Impaired STING Pathway in Human Osteosarcoma U2OS Cells Contributes to the Growth of ICP0-Null Mutant Herpes Simplex Virus. J. Virol. 2017, 91, e00006-17. [Google Scholar] [CrossRef] [Green Version]
- Sandri-Goldin, R.M. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 1998, 12, 868–879. [Google Scholar] [CrossRef]
- McCarthy, A.M.; McMahan, L.; Schaffer, P.A. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 1989, 63, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Wadd, S.E.; Lamond, A.I.; Silverstein, S.J.; Clements, J.B. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J. Virol. 2001, 75, 4376–4385. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets TBK1-activated STING singalsome to inhibit virus-induced type 1 IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef] [Green Version]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic Cleavage of VP1–2 Is Required for Release of Herpes Simplex Virus 1 DNA into the Nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-kappaB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef]
- Desai, P.; Sexton, G.L.; McCaffery, J.M.; Person, S. A Null Mutation in the Gene Encoding the Herpes Simplex Virus Type 1 UL37 Polypeptide Abrogates Virus Maturation. J. Virol. 2001, 75, 10259–10271. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.H.; Cunningham, A.L.; Diefenbach, R.J. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 2010, 84, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe 2018, 24, 234–248.e235. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [Green Version]
- Kwong, A.D.; Frenkel, N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 1926–1930. [Google Scholar] [CrossRef] [Green Version]
- Everly, D.N., Jr.; Feng, P.; Mian, I.S.; Read, G.S. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: Genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 2002, 76, 8560–8571. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, R.; Musarra-Pizzo, M.; Lei, Z.; Zhou, G.G.; Sciortino, M.T. VHS, US3 and UL13 viral tegument proteins are required for Herpes Simplex Virus-Induced modification of protein kinase R. Sci. Rep. 2020, 10, 5580. [Google Scholar] [CrossRef] [Green Version]
- Esclatine, A.; Taddeo, B.; Evans, L.; Roizman, B. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. USA 2004, 101, 3603–3608. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Zheng, S.; Huang, Z.; Lin, Y.; Shen, Q.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein UL46 Inhibits TANK-Binding Kinase 1-Mediated Signaling. mBio 2019, 10, e00919-19. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Daikoku, T.; Goshima, F.; Kume, H.; Yamaki, K.; Nishiyama, Y. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 2000, 145, 2149–2162. [Google Scholar] [CrossRef]
- Kopp, M.; Klupp, B.G.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Identification and Characterization of the Pseudorabies Virus Tegument Proteins UL46 and UL47: Role for UL47 in Virion Morphogenesis in the Cytoplasm. J. Virol. 2002, 76, 8820–8833. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J. Virol. 2017, 91, e00535-17. [Google Scholar] [CrossRef] [Green Version]
- Elliott, G.; O’Hare, P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar] [CrossRef] [Green Version]
- Kotsakis, A.; Pomeranz, L.E.; Blouin, A.; Blaho, J.A. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 2001, 75, 8697–8711. [Google Scholar] [CrossRef] [Green Version]
- Sciortino, M.T.; Taddeo, B.; Poon, A.P.; Mastino, A.; Roizman, B. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. USA 2002, 99, 8318–8323. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol. Cell 2021, 81, 2823–2837.e9. [Google Scholar] [CrossRef]
- Koshizuka, T.; Goshima, F.; Takakuwa, H.; Nozawa, N.; Daikoku, T.; Koiwai, O.; Nishiyama, Y. Identification and Characterization of the UL56 Gene Product of Herpes Simplex Virus Type 2. J. Virol. 2002, 76, 11. [Google Scholar] [CrossRef] [Green Version]
- Koshizuka, T.; Kawaguchi, Y.; Goshima, F.; Mori, I.; Nishiyama, Y. Association of Two Membrane Proteins Encoded by Herpes Simplex Virus Type 2, UL11 and UL56. Virus Genes 2006, 32, 153–163. [Google Scholar] [CrossRef]
- Zheng, Z.-Q.; Fu, Y.-Z.; Wang, S.-Y.; Xu, Z.-S.; Zou, H.-M.; Wang, Y.-Y. Herpes simplex virus protein UL56 inhibits cGAS-Mediated DNA sensing to evade antiviral immunity. Cell Insight 2022, 1, 100014. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kern, E.R.; Chatterjee, S.; Chou, J.; Roizman, B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Investig. 1993, 91, 2837–2843. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of Herpes Simplex Virus-1 Neurovirulence to γ134.5, a Gene Nonessential for Growth in Culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 1991, 72 Pt 3, 631–639. [Google Scholar] [CrossRef]
- Chou, J.; Roizman, B. The terminal a sequence of the herpes simplex virus genome contains the promoter of a gene located in the repeat sequences of the L component. J. Virol. 1986, 57, 629–637. [Google Scholar] [CrossRef] [Green Version]
- McKay, E.M.; McVey, B.; Marsden, H.S.; Brown, S.M.; MacLean, A.R. The herpes simplex virus type 1 strain 17 open reading frame RL1 encodes a polypeptide of apparent Mr 37K equivalent to ICP34.5 of herpes simplex virus type 1 strain F. J. Gen. Virol. 1993, 74, 2493–2497. [Google Scholar] [CrossRef]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3266–3270. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gross, M.; Roizman, B. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 1998, 273, 20737–20743. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Yan, Z.; Ma, Y.; Cao, Y.; He, B. A Herpesvirus Virulence Factor Inhibits Dendritic Cell Maturation through Protein Phosphatase 1 and IκB Kinase. J. Virol. 2011, 85, 3397–3407. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; Voss, K.; van Gent, M.; Chan, Y.K.; Gack, M.U.; Gale, M.; He, B. The herpesvirus accessory protein γ134.5 facilitates viral replication by disabling mitochondrial translocation of RIG-I. PLoS Pathog. 2021, 17, e1009446. [Google Scholar] [CrossRef]
- Liu, X.; Acharya, D.; Krawczyk, E.; Kangas, C.; Gack, M.U.; He, B. Herpesvirus-mediated stabilization of ICP0 expression neutralizes restriction by TRIM23. Proc. Natl. Acad. Sci. USA 2021, 118, e2113060118. [Google Scholar] [CrossRef]
- Parker, Z.M.; Murphy, A.A.; Leib, D.A. Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. J. Virol. 2015, 89, 11080–11091. [Google Scholar] [CrossRef] [Green Version]
- Royer, D.J.; Carr, D.J. A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal. Immunol. 2016, 9, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Royer, D.J.; Conrady, C.D.; Carr, D.J. Herpesvirus-Associated Lymphadenitis Distorts Fibroblastic Reticular Cell Microarchitecture and Attenuates CD8 T Cell Responses to Neurotropic Infection in Mice Lacking the STING-IFNα/β Defense Pathways. J. Immunol. 2016, 197, 2338–2352. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Dobbs, N.; Yang, K.; Yan, N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020, 53, 115–126.e115. [Google Scholar] [CrossRef]
- Skouboe, M.K.; Knudsen, A.; Reinert, L.S.; Boularan, C.; Lioux, T.; Perouzel, E.; Thomsen, M.K.; Paludan, S.R. STING agonists enable antiviral cross-talk between human cells and confer protection against genital herpes in mice. PLoS Pathog. 2018, 14, e1006976. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Zhao, J.; Xia, H.; Xie, J.; Guo, Y.; Zhong, L.; Li, M.; Yang, Q.; Peng, C.; et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 2020, 11, 6182. [Google Scholar] [CrossRef]
- Esenboga, S.; Akal, C.; Karaatmaca, B.; Erman, B.; Dogan, S.; Orhan, D.; Boztug, K.; Ayvaz, D.; Tezcan, İ. Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature. Clin. Immunol. 2018, 197, 1–5. [Google Scholar] [CrossRef]
- Murphy, A.A.; Rosato, P.C.; Parker, Z.M.; Khalenkov, A.; Leib, D.A. Synergistic control of herpes simplex virus pathogenesis by IRF-3, and IRF-7 revealed through non-invasive bioluminescence imaging. Virology 2013, 444, 71–79. [Google Scholar] [CrossRef]
- Herman, M.; Ciancanelli, M.; Ou, Y.-H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; Pérez de Diego, R.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- Andersen, L.L.; Mørk, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jørgensen, S.E.; Skipper, K.A.; Höning, K.; Gad, H.H.; Østergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczyk, E.; Kangas, C.; He, B. HSV Replication: Triggering and Repressing STING Functionality. Viruses 2023, 15, 226. https://doi.org/10.3390/v15010226
Krawczyk E, Kangas C, He B. HSV Replication: Triggering and Repressing STING Functionality. Viruses. 2023; 15(1):226. https://doi.org/10.3390/v15010226
Chicago/Turabian StyleKrawczyk, Eric, Chase Kangas, and Bin He. 2023. "HSV Replication: Triggering and Repressing STING Functionality" Viruses 15, no. 1: 226. https://doi.org/10.3390/v15010226
APA StyleKrawczyk, E., Kangas, C., & He, B. (2023). HSV Replication: Triggering and Repressing STING Functionality. Viruses, 15(1), 226. https://doi.org/10.3390/v15010226