
How Different Pathologies Are Affected by IFIT Expression



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Interferons (IFNs)
1.2. Pattern Recognition Receptors (PRRs)
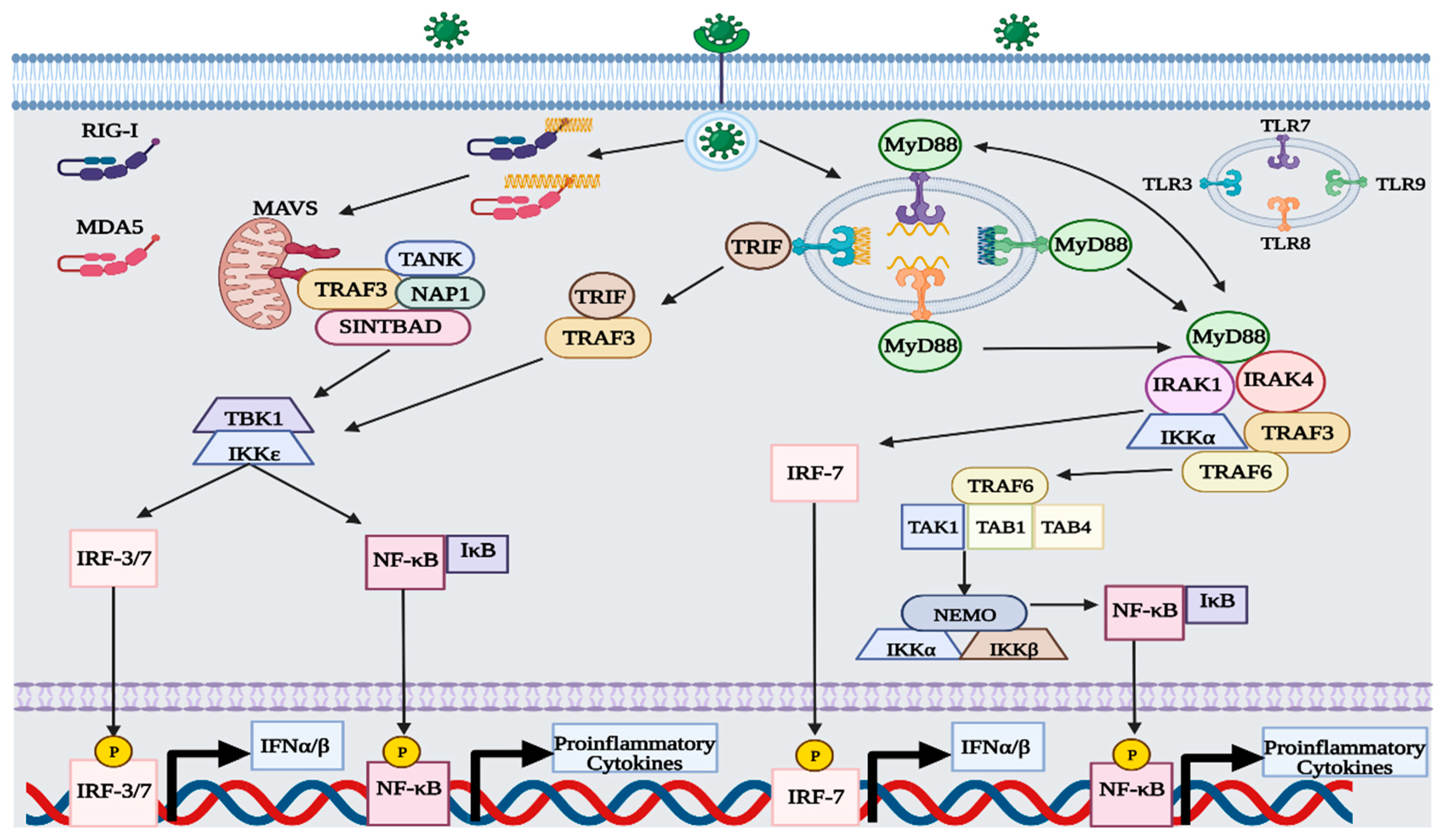
1.3. RIG-I-like Receptor (RLR) Signaling
1.4. Toll-like Receptor Signaling
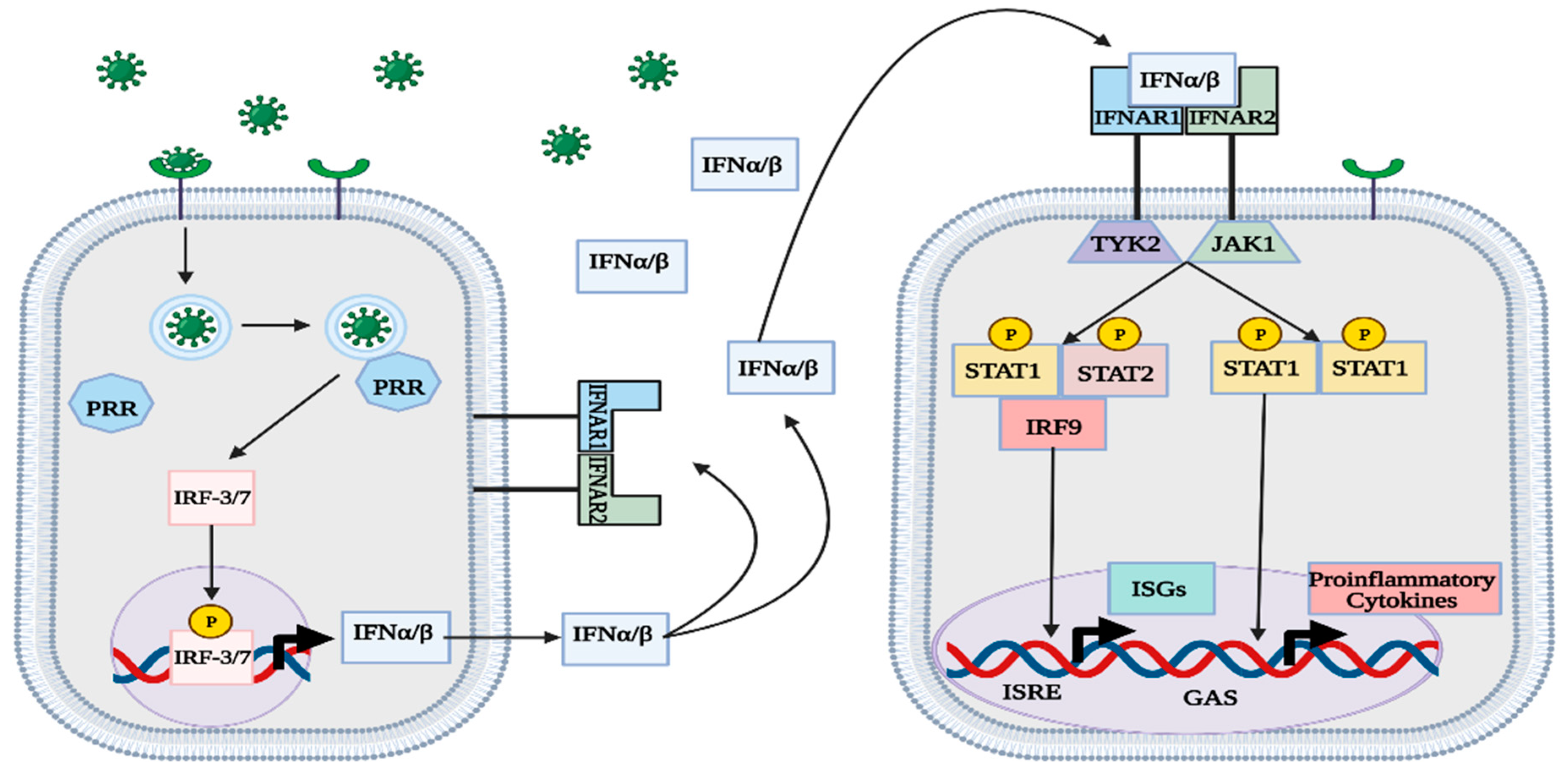
1.5. IFN Signaling
1.6. IFN-Stimulated Genes (ISGs)
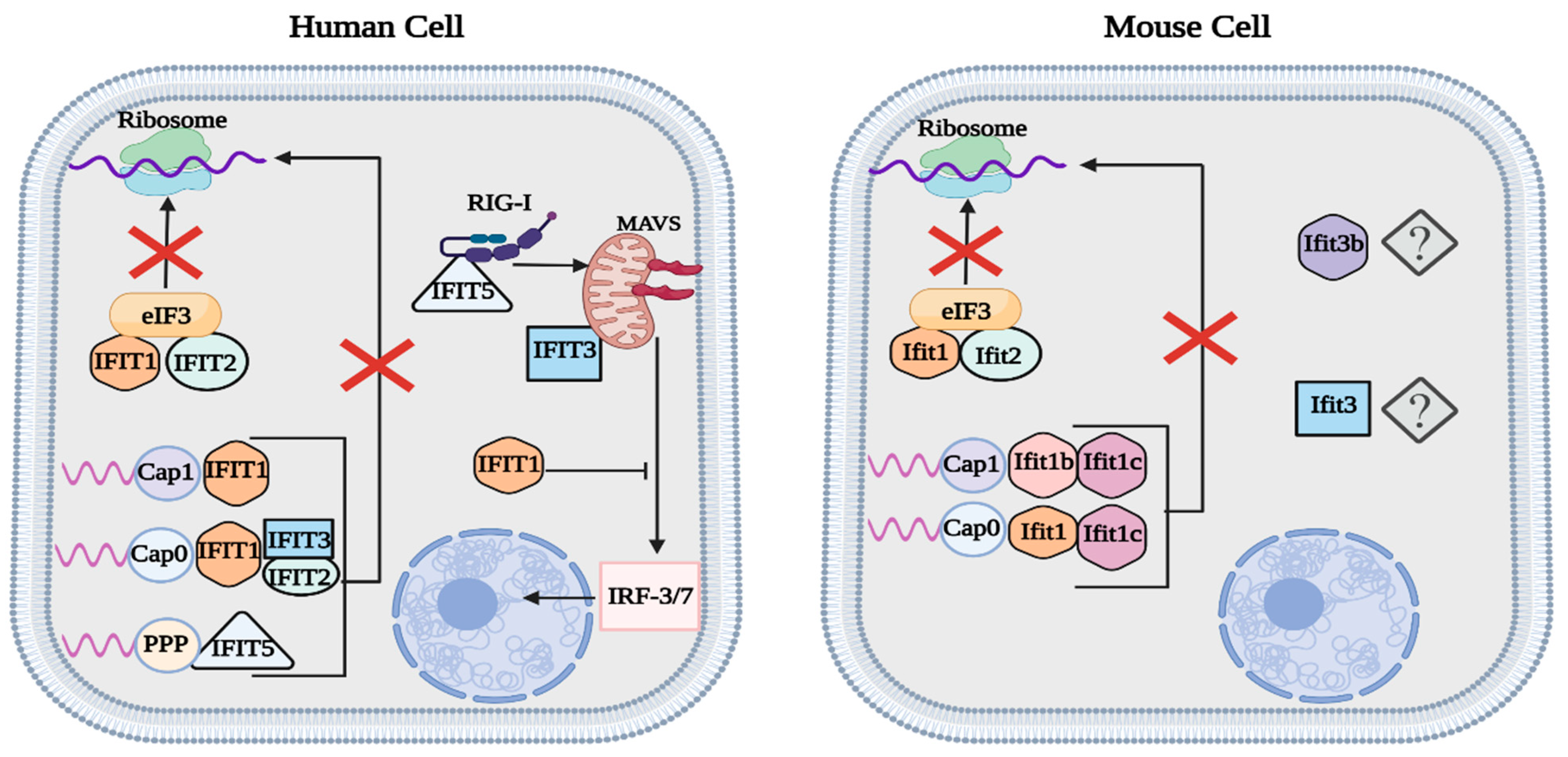
2. IFN-Induced Tetratricopeptide Repeat (IFIT) Proteins
2.1. Expression of IFITs during Viral Infection
2.2. Antiviral Functions of IFIT Proteins
3. IFIT Expression during Cancer
4. IFIT Expression during Sepsis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target 2021, 6, 291. [Google Scholar] [CrossRef]
- Levy, D.E.; Marie, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [Green Version]
- Samanta, M.; Iwakiri, D.; Takada, K. Epstein-Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 2008, 27, 4150–4160. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 2021, 13, 575. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Popli, S.; Chakravarty, S.; Fan, S.; Glanz, A.; Aras, S.; Nagy, L.E.; Sen, G.C.; Chakravarti, R.; Chattopadhyay, S. IRF3 inhibits nuclear translocation of NF-kappaB to prevent viral inflammation. Proc. Natl. Acad. Sci. USA 2022, 119, e2121385119. [Google Scholar] [CrossRef]
- Brencicova, E.; Diebold, S.S. Nucleic acids and endosomal pattern recognition: How to tell friend from foe? Front. Cell Infect. Microbiol. 2013, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Yamashita, M.; Sen, G.C. Antiviral actions of double-stranded RNA. In Nucleic Acid Sensors and Antiviral Immunity; Sambhara, S., Fujita, T., Eds.; Landes Bioscience: Austin, TX, USA, 2012. [Google Scholar]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Sen, G.C. Tyrosine phosphorylation in Toll-like receptor signaling. Cytokine Growth Factor Rev. 2014, 25, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Sen, G.C. dsRNA-activation of TLR3 and RLR signaling: Gene induction-dependent and independent effects. J. Interferon Cytokine Res. 2014, 34, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef]
- Subramanian, G.; Kuzmanovic, T.; Zhang, Y.; Peter, C.B.; Veleeparambil, M.; Chakravarti, R.; Sen, G.C.; Chattopadhyay, S. A new mechanism of interferon’s antiviral action: Induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7. PLoS Pathog. 2018, 14, e1006877. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, G.; Popli, S.; Chakravarty, S.; Taylor, R.T.; Chakravarti, R.; Chattopadhyay, S. The interferon-inducible protein TDRD7 inhibits AMP-activated protein kinase and thereby restricts autophagy-independent virus replication. J. Biol. Chem. 2020, 295, 6811–6822. [Google Scholar] [CrossRef] [Green Version]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Morales, D.J.; Lenschow, D.J. The antiviral activities of ISG15. J. Mol. Biol. 2013, 425, 4995–5008. [Google Scholar] [CrossRef]
- Mears, H.V.; Sweeney, T.R. Better together: The role of IFIT protein-protein interactions in the antiviral response. J. Gen. Virol. 2018, 99, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon induced IFIT family genes in host antiviral defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, M.D.; Schaller, A.M.; Geballe, A.P.; Malik, H.S. Evolution-guided functional analyses reveal diverse antiviral specificities encoded by IFIT1 genes in mammals. Elife 2016, 5, e14228. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Gorna, M.W.; Superti-Furga, G. IFITs: Emerging Roles as Key Anti-Viral Proteins. Front. Immunol. 2014, 5, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fensterl, V.; Wetzel, J.L.; Ramachandran, S.; Ogino, T.; Stohlman, S.A.; Bergmann, C.C.; Diamond, M.S.; Virgin, H.W.; Sen, G.C. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012, 8, e1002712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Shrestha, B.; Sen, G.C.; Diamond, M.S. A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol. 2013, 87, 8363–8371. [Google Scholar] [CrossRef] [Green Version]
- Butchi, N.B.; Hinton, D.R.; Stohlman, S.A.; Kapil, P.; Fensterl, V.; Sen, G.C.; Bergmann, C.C. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol. 2014, 88, 1051–1064. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Y.; Chen, W.; Wei, B.; Shan, Y.F.; Wang, C. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol. 2011, 187, 2559–2568. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.; VanBlargan, L.A.; Xu, W.; White, J.P.; Shan, C.; Shi, P.Y.; Zhang, R.; Adhikari, J.; Gross, M.L.; Leung, D.W.; et al. Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity 2018, 48, 487–499.e485. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2014, 42, 3228–3245. [Google Scholar] [CrossRef] [Green Version]
- Chai, B.; Tian, D.; Zhou, M.; Tian, B.; Yuan, Y.; Sui, B.; Wang, K.; Pei, J.; Huang, F.; Wu, Q.; et al. Murine Ifit3 restricts the replication of Rabies virus both in vitro and in vivo. J. Gen. Virol. 2021, 102, 001619. [Google Scholar] [CrossRef] [PubMed]
- Mears, H.V.; Sweeney, T.R. Mouse Ifit1b is a cap1-RNA-binding protein that inhibits mouse coronavirus translation and is regulated by complexing with Ifit1c. J. Biol. Chem. 2020, 295, 17781–17801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, X.; Chen, W.; Chen, L. IFIT5 potentiates anti-viral response through enhancing innate immune signaling pathways. Acta Biochim. Biophys. Sin. 2013, 45, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 2009, 106, 7945–7950. [Google Scholar] [CrossRef] [Green Version]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.M.; Liu, C.J.; Lee, T.C. Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Rivera, C. Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11884–11894. [Google Scholar]
- Lai, K.C.; Liu, C.J.; Chang, K.W.; Lee, T.C. Depleting IFIT2 mediates atypical PKC signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene 2013, 32, 3686–3697. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.C.; Chang, K.W.; Liu, C.J.; Kao, S.Y.; Lee, T.C. IFN-induced protein with tetratricopeptide repeats 2 inhibits migration activity and increases survival of oral squamous cell carcinoma. Mol. Cancer Res. 2008, 6, 1431–1439. [Google Scholar] [CrossRef] [Green Version]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liu, S.; Xu, F.; Kong, Y.; Wan, L.; Zhang, Y.; Zhang, Z. Inhibition of Proteasome Activity Induces Aggregation of IFIT2 in the Centrosome and Enhances IFIT2-Induced Cell Apoptosis. Int. J. Biol. Sci. 2017, 13, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Pidugu, V.K.; Wu, M.M.; Yen, A.H.; Pidugu, H.B.; Chang, K.W.; Liu, C.J.; Lee, T.C. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene 2019, 38, 3232–3247. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-H.; Zhao, B.-B.; Qin, C.; Wang, Y.-Y.; Li, Z.-R.; Cao, H.-T.; Yang, X.-Y.; Zhou, X.-T.; Wang, W.-B. IFIT1 modulates the proliferation, migration and invasion of pancreatic cancer cells via Wnt/β-catenin signaling. Cell. Oncol. 2021, 44, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Niess, H.; Camaj, P.; Mair, R.; Renner, A.; Zhao, Y.; Jackel, C.; Nelson, P.J.; Jauch, K.W.; Bruns, C.J. Overexpression of IFN-induced protein with tetratricopeptide repeats 3 (IFIT3) in pancreatic cancer: Cellular "pseudoinflammation" contributing to an aggressive phenotype. Oncotarget 2015, 6, 3306–3318. [Google Scholar] [CrossRef] [Green Version]
- Popp, M.C.; Klippstein, M.; Lohneis, P.; Kalinski, T.; Li, J.; Quaas, A.; Bludau, M.; Wang, Z.; Waldschmidt, D.; Kunzmann, V.; et al. Interferon-Induced Protein with Multiple Tetratricopeptide Repeats 3 Is Associated with Response to Chemotherapy and Recurrence but Not with Survival. Pancreas 2020, 49, 1307–1314. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, J.; Zhao, J.; Li, J.; Li, D.; Popp, M.; Popp, F.; Alakus, H.; Kong, B.; Dong, Q.; et al. Inflammatory IFIT3 renders chemotherapy resistance by regulating post-translational modification of VDAC2 in pancreatic cancer. Theranostics 2020, 10, 7178–7192. [Google Scholar] [CrossRef]
- Huang, J.; Lo, U.G.; Wu, S.; Wang, B.; Pong, R.C.; Lai, C.H.; Lin, H.; He, D.; Hsieh, J.T.; Wu, K. The roles and mechanism of IFIT5 in bladder cancer epithelial-mesenchymal transition and progression. Cell Death Dis. 2019, 10, 437. [Google Scholar] [CrossRef] [Green Version]
- Lo, U.G.; Pong, R.C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFNgamma-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019, 79, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.H.; Chen, X.; Pan, Z.K. Novel Treatments Targeting the Dysregulated Cell Signaling Pathway during Sepsis. J. Cell Signal 2021, 2, 228–234. [Google Scholar]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med. 2019, 7, 2050312119835043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolin, H.H.; Papadimos, T.J.; Chen, X.; Pan, Z.K. Characterization of Pathogenic Sepsis Etiologies and Patient Profiles: A Novel Approach to Triage and Treatment. Microbiol. Insights 2019, 12, 1178636118825081. [Google Scholar] [CrossRef] [Green Version]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, S.; Manncke, B.; Klenk, J.; Geisel, J.; Autenrieth, I.B.; Bohn, E. Forced IFIT-2 expression represses LPS induced TNF-alpha expression at posttranscriptional levels. BMC Immunol. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Siegfried, A.; Berchtold, S.; Manncke, B.; Deuschle, E.; Reber, J.; Ott, T.; Weber, M.; Kalinke, U.; Hofer, M.J.; Hatesuer, B.; et al. IFIT2 is an effector protein of type I IFN-mediated amplification of lipopolysaccharide (LPS)-induced TNF-alpha secretion and LPS-induced endotoxin shock. J. Immunol. 2013, 191, 3913–3921. [Google Scholar] [CrossRef] [Green Version]
- Stawowczyk, M.; Naseem, S.; Montoya, V.; Baker, D.P.; Konopka, J.; Reich, N.C. Pathogenic Effects of IFIT2 and Interferon-β during Fatal Systemic Candida albicans Infection. Mbio 2018, 9, e00365-18. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.L.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and Immune Pathogenesis of Viral Sepsis. Front Immunol 2018, 9, 2147. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Shaath, H.; Alajez, N. Protein Coding and Long Noncoding RNA (lncRNA) Transcriptional Landscape in SARS-CoV-2 Infected Bronchial Epithelial Cells Highlight a Role for Interferon and Inflammatory Response. Genes 2020, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, J.H.; Chattopadhyay, S.; Pan, Z.K. How Different Pathologies Are Affected by IFIT Expression. Viruses 2023, 15, 342. https://doi.org/10.3390/v15020342
Franco JH, Chattopadhyay S, Pan ZK. How Different Pathologies Are Affected by IFIT Expression. Viruses. 2023; 15(2):342. https://doi.org/10.3390/v15020342
Chicago/Turabian StyleFranco, Justin H., Saurabh Chattopadhyay, and Zhixing K. Pan. 2023. "How Different Pathologies Are Affected by IFIT Expression" Viruses 15, no. 2: 342. https://doi.org/10.3390/v15020342
APA StyleFranco, J. H., Chattopadhyay, S., & Pan, Z. K. (2023). How Different Pathologies Are Affected by IFIT Expression. Viruses, 15(2), 342. https://doi.org/10.3390/v15020342