
HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Reagents
2.2. Generation of Virus Stocks and Molecular Clones
2.3. Determination of TCID50 Values for Wildtype and Mutant HIV Stocks and Molecular Clones
2.4. Serial Passage for Selection of Resistant Virus
2.5. Sequencing of Passaged Virus
2.6. Enzyme-Linked Immunosorbent Assay (ELISA) to Determine p24 Levels in Viral Stocks
2.7. Dose-Response Assays to Determine Sensitivity to Antivirals
2.8. Determination of Specific Infectivity
2.9. Endogenous Reverse Transcriptase Assay
2.10. Statistics
3. Results
3.1. Resistance Mutations Found during Serial Passages of WT, K65R, M184V Viruses in the Presence of ISL and TDF
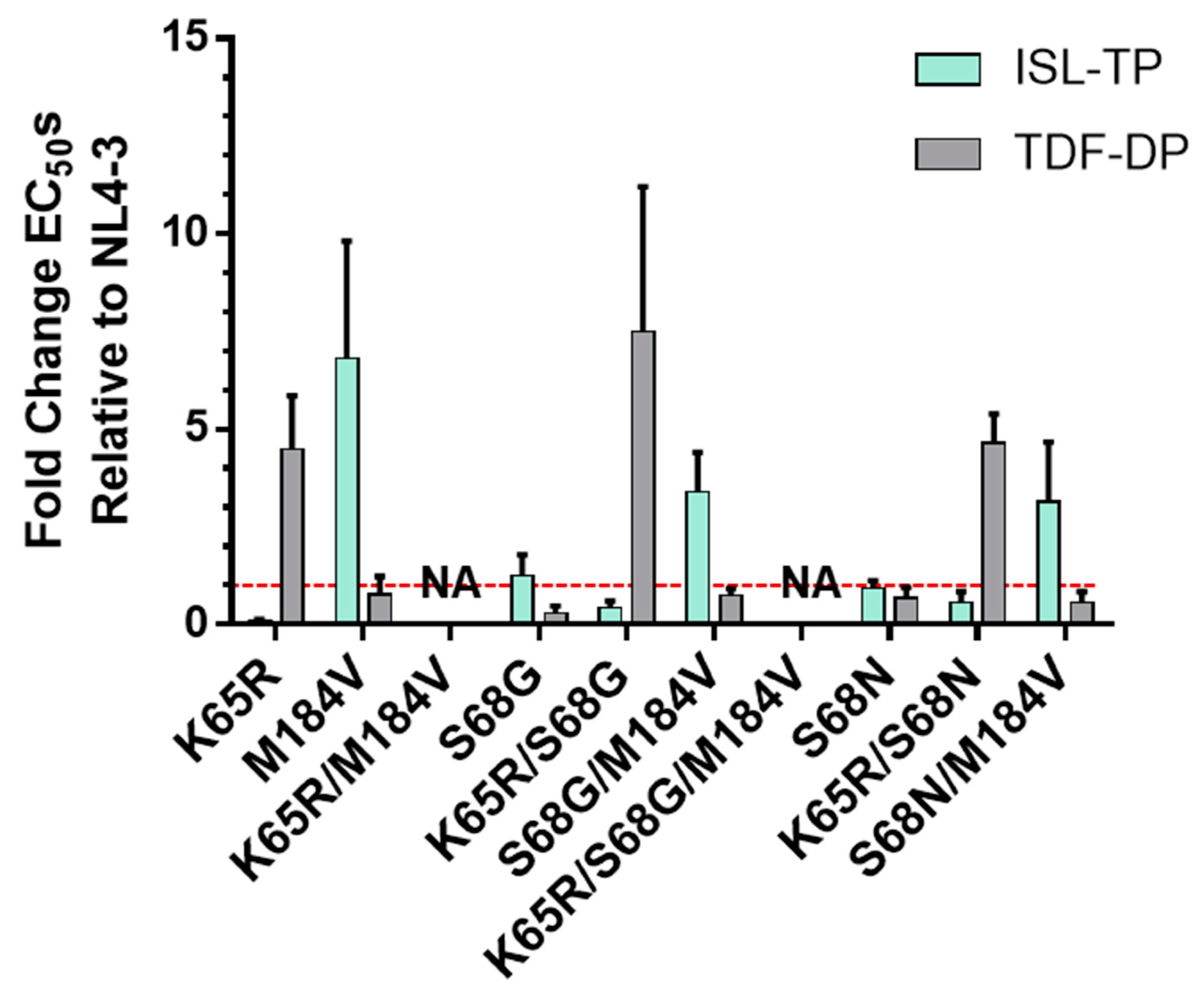
3.2. Validation of Mutations Found in TDF- and ISL-Passaging Using Molecular Clones and Evaluating the Specific Infectivity
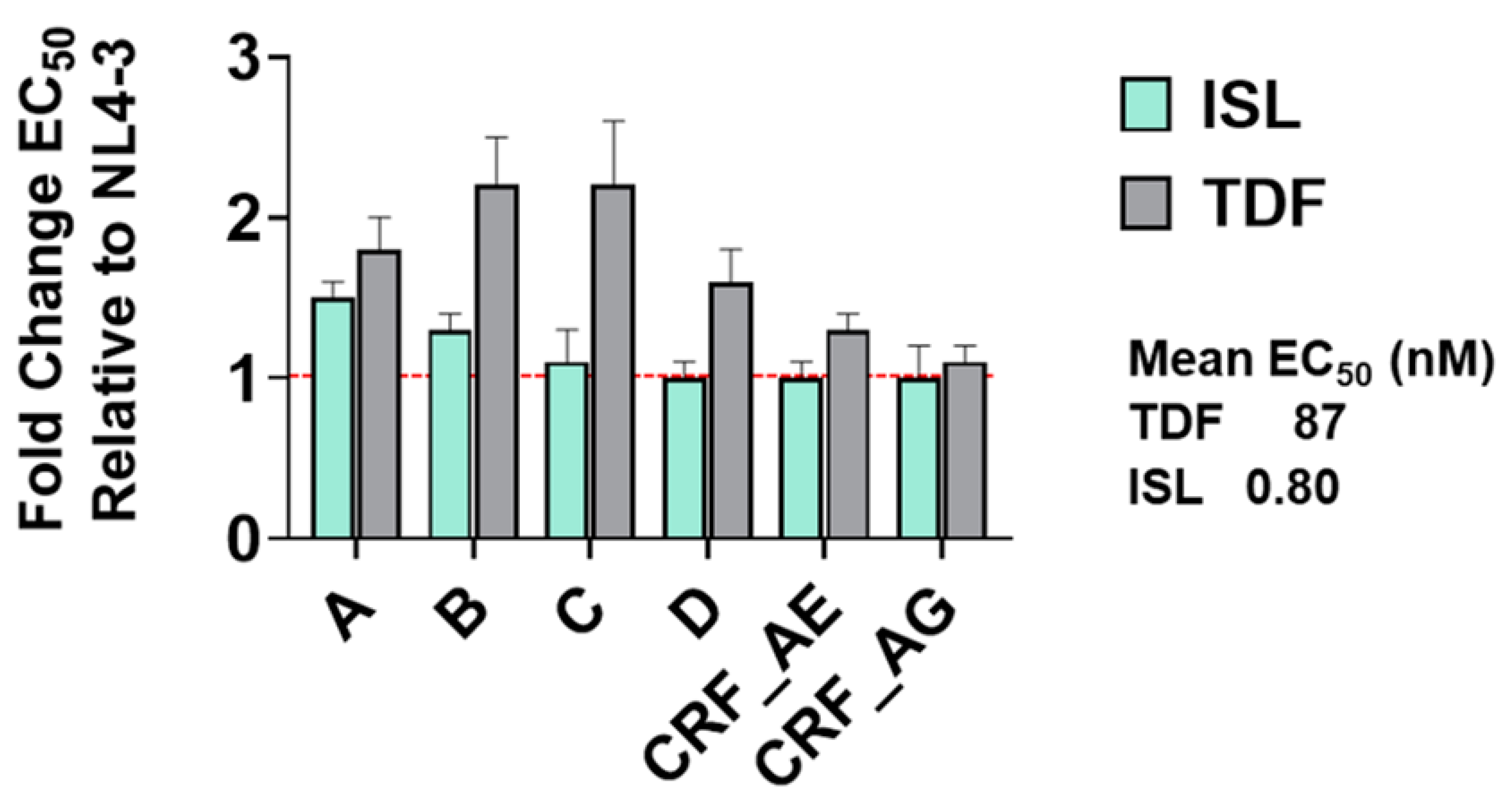
3.3. Potency of ISL and TDF against Diverse HIV Subtypes
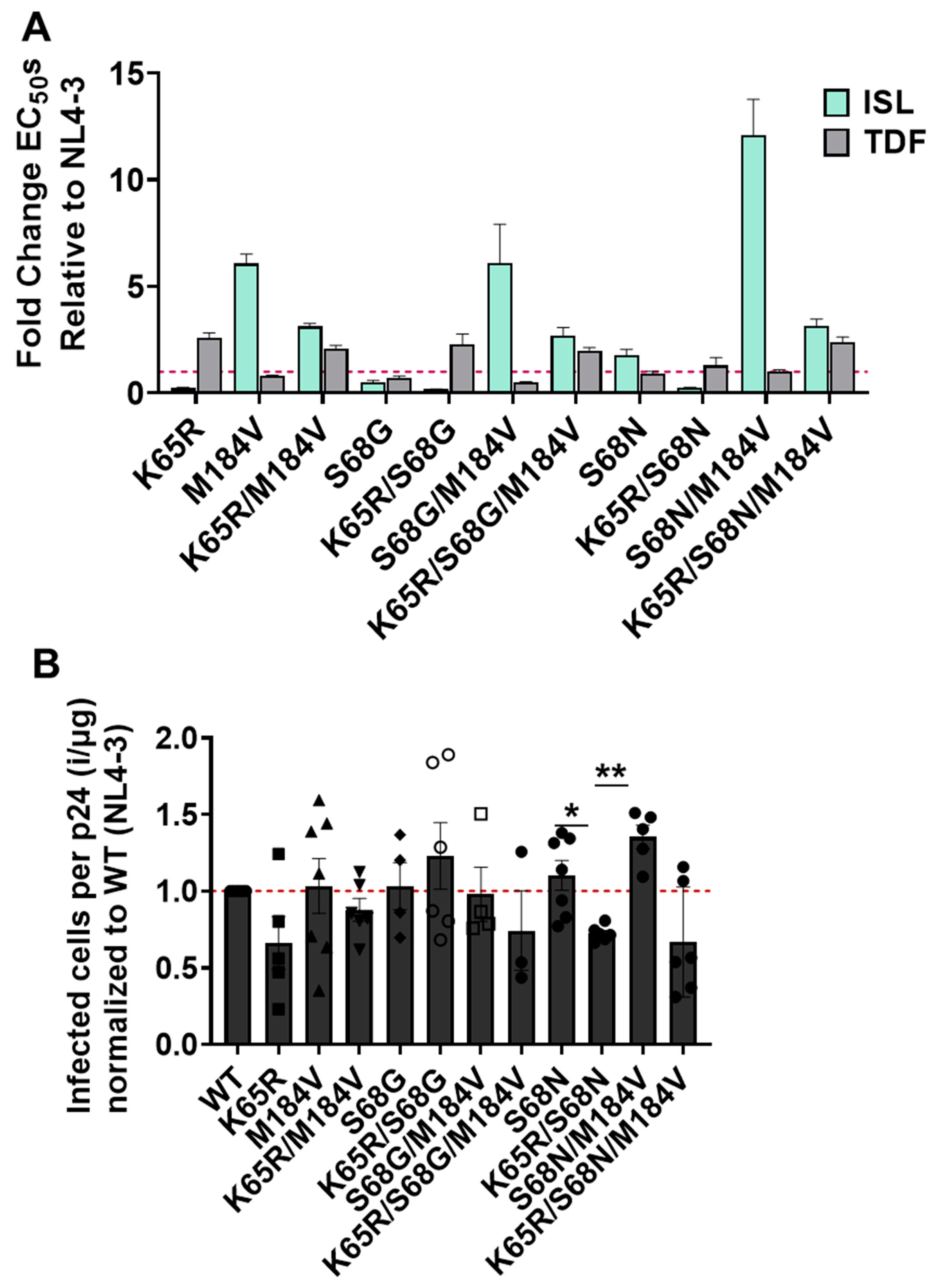
3.4. Impact of Mutations K65R, M184V, and K65R/M184V on Susceptibility to ISL and TDF and Viral Fitness
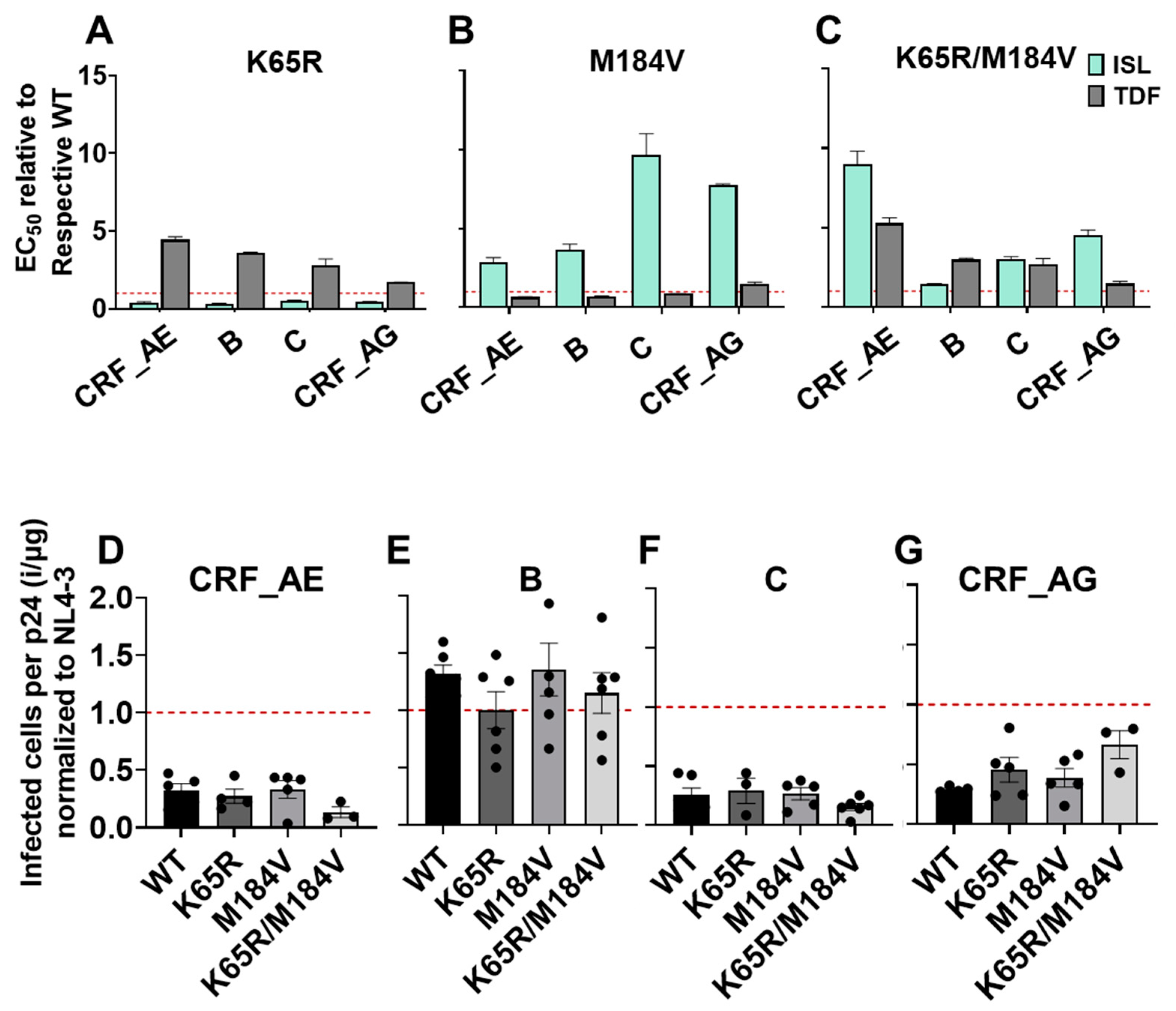
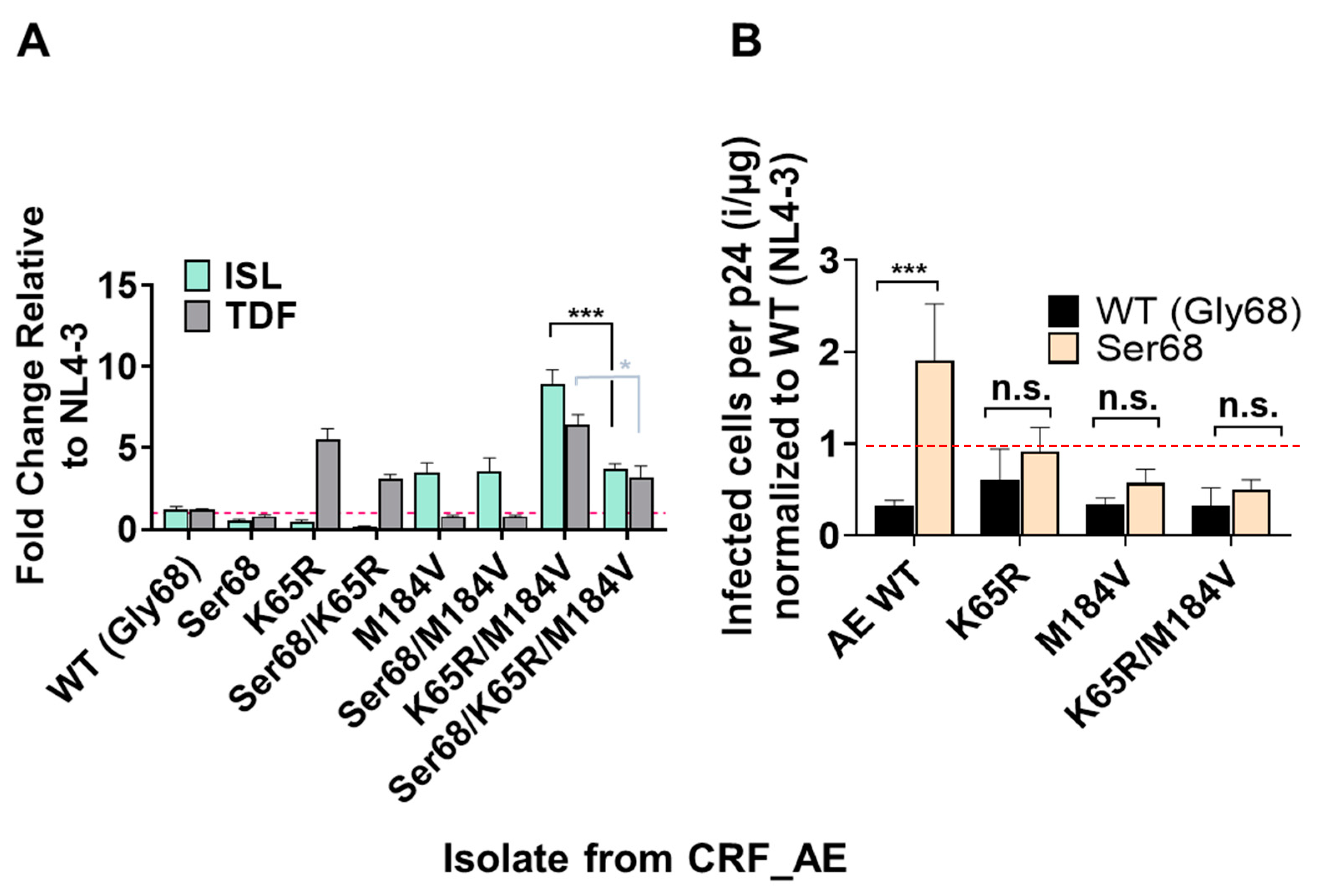
3.5. Polymorphism of Gly68 in CRF_AE K65R/M184V Influences Susceptibility to ISL and TFV and Viral Fitness
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. Aids 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.K.; Salminen, M.O.; Koch, C.; Gotte, D.; Artenstein, A.W.; Hegerich, P.A.; St Louis, D.; Burke, D.S.; McCutchan, F.E. Full-length sequence and mosaic structure of a human immunodeficiency virus type 1 isolate from Thailand. J. Virol. 1996, 70, 5935–5943. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Robertson, D.L.; Morrison, S.G.; Hui, H.; Craig, S.; Decker, J.; Fultz, P.N.; Girard, M.; Shaw, G.M.; Hahn, B.H.; et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J. Virol. 1996, 70, 7013–7029. [Google Scholar] [CrossRef] [PubMed]
- McCutchan, F.E.; Hegerich, P.A.; Brennan, T.P.; Phanuphak, P.; Singharaj, P.; Jugsudee, A.; Berman, P.W.; Gray, A.M.; Fowler, A.K.; Burke, D.S. Genetic variants of HIV-1 in Thailand. AIDS Res. Hum. Retrovir. 1992, 8, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Fettig, J.; Swaminathan, M.; Murrill, C.S.; Kaplan, J.E. Global epidemiology of HIV. Infect. Dis. Clin. N. Am. 2014, 28, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; Patel, D.; Ehteshami, M. The best backbone for HIV prevention, treatment, and elimination: Emtricitabine+tenofovir. Antivir. Ther. 2022, 27, 13596535211067599. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Wu, H.Y.; Yarla, N.S.; Xu, B.; Ding, J.; Lu, T.R. HAART in HIV/AIDS Treatments: Future Trends. Infect. Disord. Drug Targets 2018, 18, 15–22. [Google Scholar] [CrossRef]
- Michailidis, E.; Huber, A.D.; Ryan, E.M.; Ong, Y.T.; Leslie, M.D.; Matzek, K.B.; Singh, K.; Marchand, B.; Hagedorn, A.N.; Kirby, K.A.; et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol. Chem. 2014, 289, 24533–24548. [Google Scholar] [CrossRef]
- Theys, K.; Vercauteren, J.; Snoeck, J.; Zazzi, M.; Camacho, R.J.; Torti, C.; Schülter, E.; Clotet, B.; Sönnerborg, A.; De Luca, A.; et al. HIV-1 subtype is an independent predictor of reverse transcriptase mutation K65R in HIV-1 patients treated with combination antiretroviral therapy including tenofovir. Antimicrob. Agents Chemother. 2013, 57, 1053–1056. [Google Scholar] [CrossRef]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; et al. 2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Murphey-Corb, M.; Rajakumar, P.; Michael, H.; Nyaundi, J.; Didier, P.J.; Reeve, A.B.; Mitsuya, H.; Sarafianos, S.G.; Parniak, M.A. Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine in vitro and in vivo. Antimicrob. Agents Chemother. 2012, 56, 4707–4712. [Google Scholar] [CrossRef] [PubMed]
- Ohrui, H.; Kohgo, S.; Hayakawa, H.; Kodama, E.; Matsuoka, M.; Nakata, T.; Mitsuya, H. 2′-Deoxy-4′-C-Ethynyl-2-Fluoroadenosine: A Nucleoside Reverse Transcriptase Inhibitor with Highly Potent Activity Against Wide Spectrum of HIV-1 Strains, Favorable Toxic Profiles, and Stability in Plasma. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1543–1546. [Google Scholar] [CrossRef]
- Stoddart, C.A.; Galkina, S.A.; Joshi, P.; Kosikova, G.; Moreno, M.E.; Rivera, J.M.; Sloan, B.; Reeve, A.B.; Sarafianos, S.G.; Murphey-Corb, M.; et al. Oral administration of the nucleoside EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob. Agents Chemother. 2015, 59, 4190–4198. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.M.; Yazdanpanah, Y.; Afani Saud, A.; Bettacchi, C.; Chahin Anania, C.; Klopfer, S.O.; Grandhi, A.; Eves, K.; Hepler, D.; Robertson, M.N.; et al. Brief Report: Efficacy and Safety of Oral Islatravir Once Daily in Combination With Doravirine Through 96 Weeks for Treatment-Naive Adults With HIV-1 Infection Receiving Initial Treatment With Islatravir, Doravirine, and Lamivudine. J. Acquir. Immune Defic. Syndr. 2022, 91, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Merck & Co. Merck Provides Update on Phase 2 Clinical Trial of Once-Weekly Investigational Combination of MK-8507 and Islatravir for the Treatment of People Living with HIV-1. 2021. Available online: https://www.merck.com/news/merck-provides-update-on-phase-2-clinical-trial-of-once-weekly-investigational-combination-of-mk-8507-and-islatravir-for-the-treatment-of-people-living-with-hiv-1/ (accessed on 18 November 2021).
- Merck & Co. Merck to Initiate New Phase 3 Clinical Program with Lower Dose of Daily Oral Islatravir in Combination with Doravirine for Treatment of People with HIV-1 Infection. 2022. Available online: https://www.merck.com/news/merck-to-initiate-new-phase-3-clinical-program-with-lower-dose-of-daily-oral-islatravir-in-combination-with-doravirine-for-treatment-of-people-with-hiv-1-infection/ (accessed on 20 September 2022).
- Kirby, K.A.; Michailidis, E.; Fetterly, T.L.; Steinbach, M.A.; Singh, K.; Marchand, B.; Leslie, M.D.; Hagedorn, A.N.; Kodama, E.N.; Marquez, V.E.; et al. Effects of substitutions at the 4′ and 2 positions on the bioactivity of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 2013, 57, 6254–6264. [Google Scholar] [CrossRef] [PubMed]
- Salie, Z.L.; Kirby, K.A.; Michailidis, E.; Marchand, B.; Singh, K.; Rohan, L.C.; Kodama, E.N.; Mitsuya, H.; Parniak, M.A.; Sarafianos, S.G. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. USA 2016, 113, 9274–9279. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Marchand, B.; Kodama, E.N.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Feng, J.Y.; Margot, N.A.; Myrick, F.; Goodman, D.; Ly, J.K.; White, K.L.; Kutty, N.; Wang, R.; Borroto-Esoda, K.; et al. The A62V and S68G mutations in HIV-1 reverse transcriptase partially restore the replication defect associated with the K65R mutation. J. Acquir. Immune Defic. Syndr. 2008, 48, 428–436. [Google Scholar] [CrossRef]
- Charneau, P.; Mirambeau, G.; Roux, P.; Paulous, S.; Buc, H.; Clavel, F. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 1994, 241, 651–662. [Google Scholar] [CrossRef]
- Haertle, T.; Carrera, C.J.; Wasson, D.B.; Sowers, L.C.; Richman, D.D.; Carson, D.A. Metabolism and anti-human immunodeficiency virus-1 activity of 2-halo-2′,3′-dideoxyadenosine derivatives. J. Biol. Chem. 1988, 263, 5870–5875. [Google Scholar] [CrossRef] [PubMed]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef] [PubMed]
- Borkow, G.; Fletcher, R.S.; Barnard, J.; Arion, D.; Motakis, D.; Dmitrienko, G.I.; Parniak, M.A. Inhibition of the ribonuclease H and DNA polymerase activities of HIV-1 reverse transcriptase by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone. Biochemistry 1997, 36, 3179–3185. [Google Scholar] [CrossRef] [PubMed]
- Motakis, D.; Parniak, M.A. A tight-binding mode of inhibition is essential for anti-human immunodeficiency virus type 1 virucidal activity of nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2002, 46, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Christensen, D.E.; Ganser-Pornillos, B.K.; Johnson, J.S.; Pornillos, O.; Sundquist, W.I. Reconstitution and visualization of HIV-1 capsid-dependent replication and integration in vitro. Science 2020, 370, eabc8420. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.; Shi, J.; Varadarajan, J.; Jamieson, P.J.; Aiken, C. The Host Cell Metabolite Inositol Hexakisphosphate Promotes Efficient Endogenous HIV-1 Reverse Transcription by Stabilizing the Viral Capsid. mBio 2020, 11, 02820. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Das, D.; Kohgo, S.; Hayashi, H.; Delino, N.S.; Sarafianos, S.G.; Mitsuya, H.; Maeda, K. The High Genetic Barrier of EFdA/MK-8591 Stems from Strong Interactions with the Active Site of Drug-Resistant HIV-1 Reverse Transcriptase. Cell Chem. Biol. 2018, 25, 1268–1278.e3. [Google Scholar] [CrossRef]
- Petrella, M.; Oliveira, M.; Moisi, D.; Detorio, M.; Brenner, B.G.; Wainberg, M.A. Differential maintenance of the M184V substitution in the reverse transcriptase of human immunodeficiency virus type 1 by various nucleoside antiretroviral agents in tissue culture. Antimicrob. Agents Chemother. 2004, 48, 4189–4194. [Google Scholar] [CrossRef]
- Michailidis, E.; Ryan, E.M.; Hachiya, A.; Kirby, K.A.; Marchand, B.; Leslie, M.D.; Huber, A.D.; Ong, Y.T.; Jackson, J.C.; Singh, K.; et al. Hypersusceptibility mechanism of Tenofovir-resistant HIV to EFdA. Retrovirology 2013, 10, 65. [Google Scholar] [CrossRef]
- Maeda, K.; Desai, D.V.; Aoki, M.; Nakata, H.; Kodama, E.N.; Mitsuya, H. Delayed emergence of HIV-1 variants resistant to 4′-ethynyl-2-fluoro-2′-deoxyadenosine: Comparative sequential passage study with lamivudine, tenofovir, emtricitabine and BMS-986001. Antivir. Ther. 2014, 19, 179–189. [Google Scholar] [CrossRef]
- Brenner, B.G.; Coutsinos, D. The K65R mutation in HIV-1 reverse transcriptase: Genetic barriers, resistance profile and clinical implications. HIV Ther. 2009, 3, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Cilento, M.E.; Reeve, A.B.; Michailidis, E.; Ilina, T.V.; Nagy, E.; Mitsuya, H.; Parniak, M.A.; Tedbury, P.R.; Sarafianos, S.G. Development of Human Immunodeficiency Virus Type 1 Resistance to 4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine Starting with Wild-Type or Nucleoside Reverse Transcriptase Inhibitor-Resistant Strains. Antimicrob. Agents Chemother. 2021, 65, e0116721. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; White, K.L.; Miller, M.D.; Parkin, N.T.; Courcambeck, J.; Halfon, P.; Selmi, B.; Boretto, J.; Canard, B. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 2004, 279, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.A.; Isaacson, E.; McGowan, I.; Cheng, A.K.; Schooley, R.T.; Miller, M.D. Genotypic and phenotypic analyses of HIV-1 in antiretroviral-experienced patients treated with tenofovir DF. AIDS 2002, 16, 1227–1235. [Google Scholar] [CrossRef]
- Miller, M.D. K65R, TAMs and tenofovir. AIDS Rev. 2004, 6, 22–33. [Google Scholar] [PubMed]
- Scherrer, A.U.; von Wyl, V.; Götte, M.; Klimkait, T.; Cellerai, C.; Yerly, S.; Böni, J.; Held, L.; Ledergerber, B.; Günthard, H.F. Polymorphic mutations associated with the emergence of the multinucleoside/tide resistance mutations 69 insertion and Q151M. J. Acquir. Immune Defic. Syndr. 2012, 59, 105–112. [Google Scholar] [CrossRef]
- Wirden, M.; Malet, I.; Derache, A.; Marcelin, A.G.; Roquebert, B.; Simon, A.; Kirstetter, M.; Joubert, L.M.; Katlama, C.; Calvez, V. Clonal analyses of HIV quasispecies in patients harbouring plasma genotype with K65R mutation associated with thymidine analogue mutations or L74V substitution. Aids 2005, 19, 630–632. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Kantor, R.; Katzenstein, D.A.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.F.; Schapiro, J.M.; Shafer, R.W. HIV-1 pol mutation frequency by subtype and treatment experience: Extension of the HIVseq program to seven non-B subtypes. Aids 2006, 20, 643–651. [Google Scholar] [CrossRef]
- Shafer, R.W. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006, 194 (Suppl. 1), S51–S58. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Jung, D.R.; Betts, B.J. Human immunodeficiency virus type 1 reverse transcriptase and protease mutation search engine for queries. Nat. Med. 2000, 6, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Chimukangara, B.; Lessells, R.J.; Rhee, S.Y.; Giandhari, J.; Kharsany, A.B.M.; Naidoo, K.; Lewis, L.; Cawood, C.; Khanyile, D.; Ayalew, K.A.; et al. Trends in Pretreatment HIV-1 Drug Resistance in Antiretroviral Therapy-naive Adults in South Africa, 2000–2016: A Pooled Sequence Analysis. EClinicalMedicine 2019, 9, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, S.; Han, Y.; Zhuang, D.; Li, L.; Liu, Y.; Liu, S.; Bao, Z.; Zhang, W.; Song, H.; et al. The prevalence of drug resistance among treatment-naïve HIV-1-infected individuals in China during pre- and post- 2004. BMC Infect. Dis. 2016, 16, 605. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, S.M.; Lee, G.Q.; Kamelian, K.; Kembabazi, A.; Musinguzi, N.; Bwana, M.B.; Muzoora, C.; Haberer, J.E.; Hunt, P.W.; Martin, J.N.; et al. Increasing Prevalence of HIV Pretreatment Drug Resistance in Women But Not Men in Rural Uganda During 2005–2013. AIDS Patient Care STDS 2018, 32, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Mega, E.R. Alarming surge in drug-resistant HIV uncovered. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Kassaye, S.G.; Barrow, G.; Sundaramurthi, J.C.; Jordan, M.R.; Shafer, R.W. HIV-1 transmitted drug resistance surveillance: Shifting trends in study design and prevalence estimates. J. Int. AIDS Soc. 2020, 23, e25611. [Google Scholar] [CrossRef]
- Bazmi, H.Z.; Hammond, J.L.; Cavalcanti, S.C.; Chu, C.K.; Schinazi, R.F.; Mellors, J.W. In vitro selection of mutations in the human immunodeficiency virus type 1 reverse transcriptase that decrease susceptibility to (-)-beta-D-dioxolane-guanosine and suppress resistance to 3′-azido-3′-deoxythymidine. Antimicrob. Agents Chemother. 2000, 44, 1783–1788. [Google Scholar] [CrossRef]
- Naeger, L.K.; Struble, K.A. Effect of baseline protease genotype and phenotype on HIV response to atazanavir/ritonavir in treatment-experienced patients. Aids 2006, 20, 847–853. [Google Scholar] [CrossRef]
- Zhang, D.; Caliendo, A.M.; Eron, J.J.; DeVore, K.M.; Kaplan, J.C.; Hirsch, M.S.; D’Aquila, R.T. Resistance to 2′,3′-dideoxycytidine conferred by a mutation in codon 65 of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1994, 38, 282–287. [Google Scholar] [CrossRef]
- Diamond, T.L.; Ngo, W.; Xu, M.; Goh, S.L.; Rodriguez, S.; Lai, M.T.; Asante-Appiah, E.; Grobler, J.A. Islatravir Has a High Barrier to Resistance and Exhibits a Differentiated Resistance Profile from Approved Nucleoside Reverse Transcriptase Inhibitors (NRTIs). Antimicrob. Agents Chemother. 2022, 66, e0013322. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Reeve, A.B.; Marchand, B.; Michailidis, E.; Ong, Y.T.; Kirby, K.A.; Leslie, M.D.; Oka, S.; Kodama, E.N.; Rohan, L.C.; et al. Evaluation of Combinations of 4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine with Clinically Used Antiretroviral Drugs. Antimicrob. Agents Chemother. 2013, 57, 4554–4558. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Clark, A.D., Jr.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 10027–10032. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; Lloyd, R.M., Jr.; Nguyen, M.H.; Cannon, D.L.; McMillan, A.; Ilksoy, N.; Chu, C.K.; Liotta, D.C.; Bazmi, H.Z.; Mellors, J.W. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob. Agents Chemother. 1993, 37, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.; Kemp, S.D.; Parry, N.R.; Larder, B.A. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3′-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. USA 1993, 90, 5653–5656. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.A.; Waters, J.M.; Miller, M.D. In vitro human immunodeficiency virus type 1 resistance selections with combinations of tenofovir and emtricitabine or abacavir and lamivudine. Antimicrob. Agents Chemother. 2006, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Røge, B.T.; Katzenstein, T.L.; Obel, N.; Nielsen, H.; Kirk, O.; Pedersen, C.; Mathiesen, L.; Lundgren, J.; Gerstoft, J. K65R with and without S68: A new resistance profile in vivo detected in most patients failing abacavir, didanosine and stavudine. Antivir. Ther. 2003, 8, 173–182. [Google Scholar] [CrossRef] [PubMed]
- García-Lerma, J.G.; Gerrish, P.J.; Wright, A.C.; Qari, S.H.; Heneine, W. Evidence of a role for the Q151L mutation and the viral background in development of multiple dideoxynucleoside-resistant human immunodeficiency virus type 1. J. Virol. 2000, 74, 9339–9346. [Google Scholar] [CrossRef]
- Kavlick, M.F.; Wyvill, K.; Yarchoan, R.; Mitsuya, H. Emergence of multi-dideoxynucleoside-resistant human immunodeficiency virus type 1 variants, viral sequence variation, and disease progression in patients receiving antiretroviral chemotherapy. J. Infect. Dis. 1998, 177, 1506–1513. [Google Scholar] [CrossRef]
- Shirasaka, T.; Kavlick, M.F.; Ueno, T.; Gao, W.Y.; Kojima, E.; Alcaide, M.L.; Chokekijchai, S.; Roy, B.M.; Arnold, E.; Yarchoan, R.; et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 1995, 92, 2398–2402. [Google Scholar] [CrossRef]
- Doualla-Bell, F.; Avalos, A.; Brenner, B.; Gaolathe, T.; Mine, M.; Gaseitsiwe, S.; Oliveira, M.; Moisi, D.; Ndwapi, N.; Moffat, H.; et al. High prevalence of the K65R mutation in human immunodeficiency virus type 1 subtype C isolates from infected patients in Botswana treated with didanosine-based regimens. Antimicrob. Agents Chemother. 2006, 50, 4182–4185. [Google Scholar] [CrossRef]
- Li, S.; Ouyang, J.; Zhao, B.; An, M.; Wang, L.; Ding, H.; Zhang, M.; Han, X. The S68G polymorphism is a compensatory mutation associated with the drug resistance mutation K65R in CRF01_AE strains. BMC Infect. Dis. 2020, 20, 123. [Google Scholar] [CrossRef]
- Brown, B.K.; Darden, J.M.; Tovanabutra, S.; Oblander, T.; Frost, J.; Sanders-Buell, E.; de Souza, M.S.; Birx, D.L.; McCutchan, F.E.; Polonis, V.R. Biologic and genetic characterization of a panel of 60 human immunodeficiency virus type 1 isolates, representing clades A, B, C, D, CRF01_AE, and CRF02_AG, for the development and assessment of candidate vaccines. J. Virol. 2005, 79, 6089–6101. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | TDF:ISL | Days of Passage | Final Passage Number | Amino Acid Mutations | Proportion of Sequence Population (%) |
|---|---|---|---|---|---|
| Wildtype | 1:1 | 34 | 6 | M184I | 71.4 |
| M184V | 28.6 | ||||
| 10:1 | 65 | 5 | M184V | 100 | |
| 100:1 | 97 | 2 | None | 90.5 | |
| M184I | 9.5 | ||||
| M184V | 1:1 | 87 | 5 | None | 81 |
| K65R | 9.5 | ||||
| M184I | 4.8 | ||||
| V184M | 4.8 | ||||
| K65R | 1:1 | 32 | 7 | M184V | 95.7 |
| S68G/M184V | 4.3 | ||||
| 10:1 | 36 | 7 | M184V | 100 | |
| 100:1 | 58 | 5 | S68N | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilento, M.E.; Wen, X.; Reeve, A.B.; Ukah, O.B.; Snyder, A.A.; Carrillo, C.M.; Smith, C.P.; Edwards, K.; Wahoski, C.C.; Kitzler, D.R.; et al. HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes. Viruses 2023, 15, 1990. https://doi.org/10.3390/v15101990
Cilento ME, Wen X, Reeve AB, Ukah OB, Snyder AA, Carrillo CM, Smith CP, Edwards K, Wahoski CC, Kitzler DR, et al. HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes. Viruses. 2023; 15(10):1990. https://doi.org/10.3390/v15101990
Chicago/Turabian StyleCilento, Maria E., Xin Wen, Aaron B. Reeve, Obiaara B. Ukah, Alexa A. Snyder, Ciro M. Carrillo, Cole P. Smith, Kristin Edwards, Claudia C. Wahoski, Deborah R. Kitzler, and et al. 2023. "HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes" Viruses 15, no. 10: 1990. https://doi.org/10.3390/v15101990
APA StyleCilento, M. E., Wen, X., Reeve, A. B., Ukah, O. B., Snyder, A. A., Carrillo, C. M., Smith, C. P., Edwards, K., Wahoski, C. C., Kitzler, D. R., Kodama, E. N., Mitsuya, H., Parniak, M. A., Tedbury, P. R., & Sarafianos, S. G. (2023). HIV-1 Resistance to Islatravir/Tenofovir Combination Therapy in Wild-Type or NRTI-Resistant Strains of Diverse HIV-1 Subtypes. Viruses, 15(10), 1990. https://doi.org/10.3390/v15101990