

Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites



Abstract
:1. Introduction
2. Materials and Methods
2.1. Structural Modeling and Refinement
2.2. Coarse-Grained Dynamics Simulations
2.3. All-Atom Molecular Dynamics Simulations
2.4. Network Analysis of Conformational Ensembles
2.5. Machine Learning-Based Identification of Cryptic Pockets and Network-Based Ranking of Allosteric Pocket Propensities and Allosteric Binding Sites
3. Results
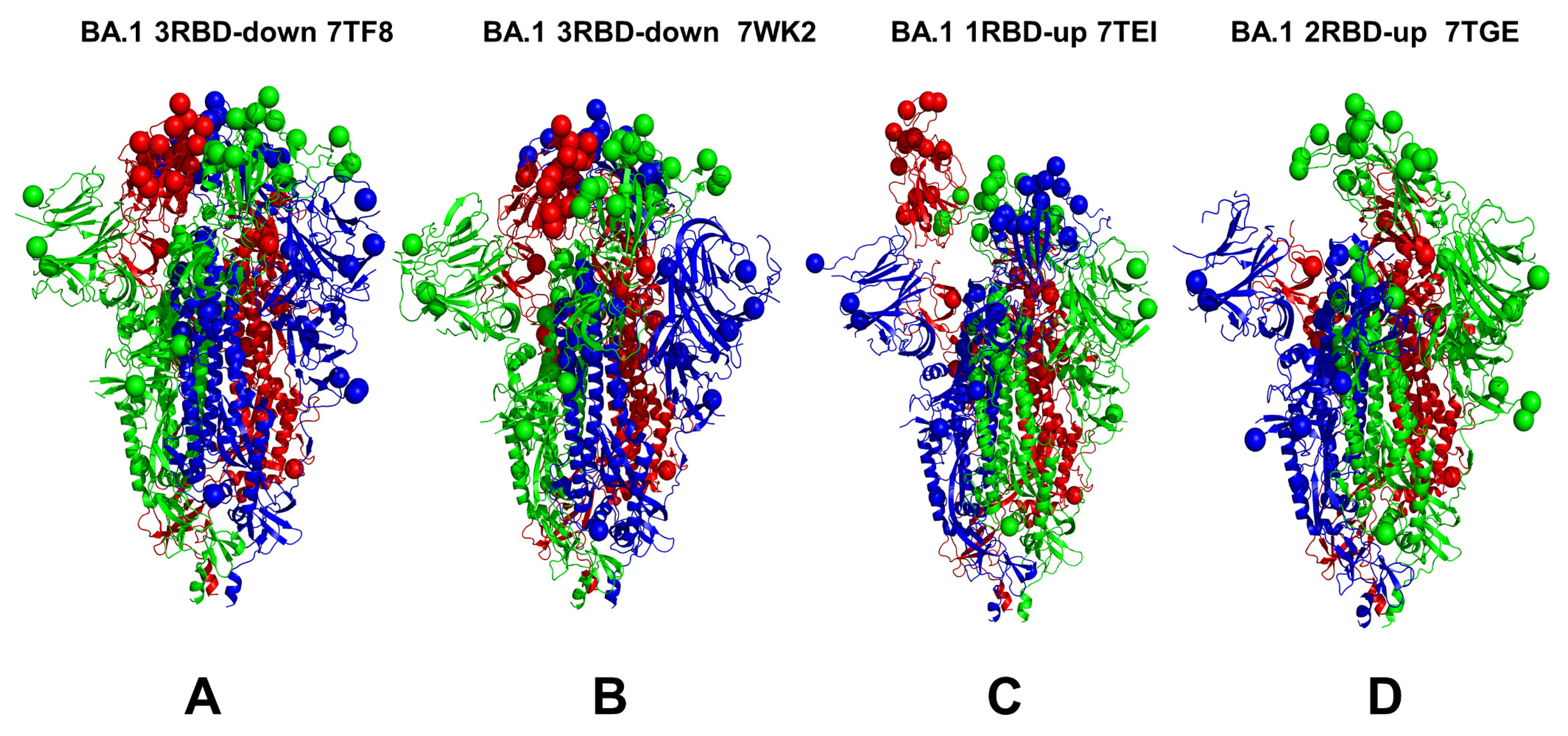
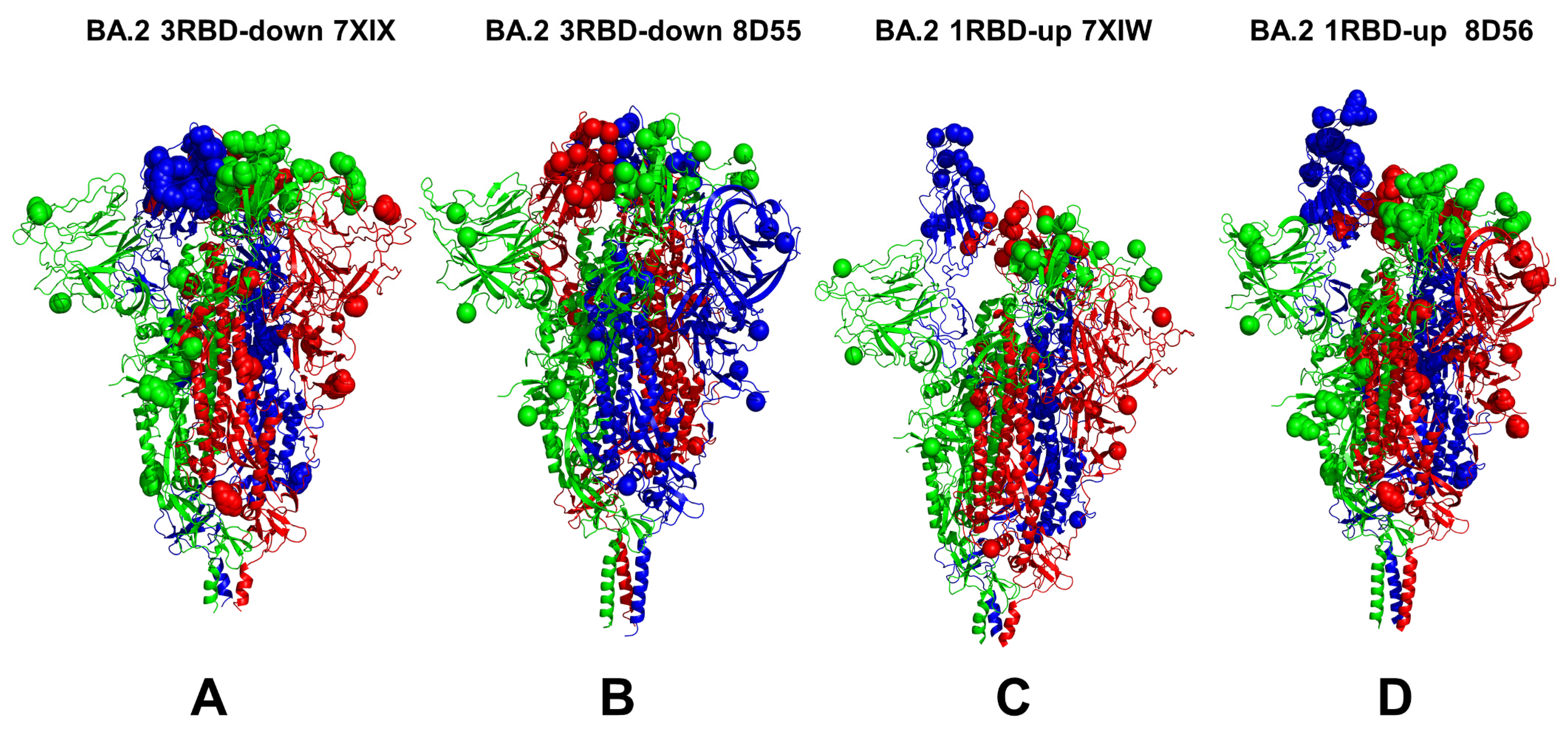
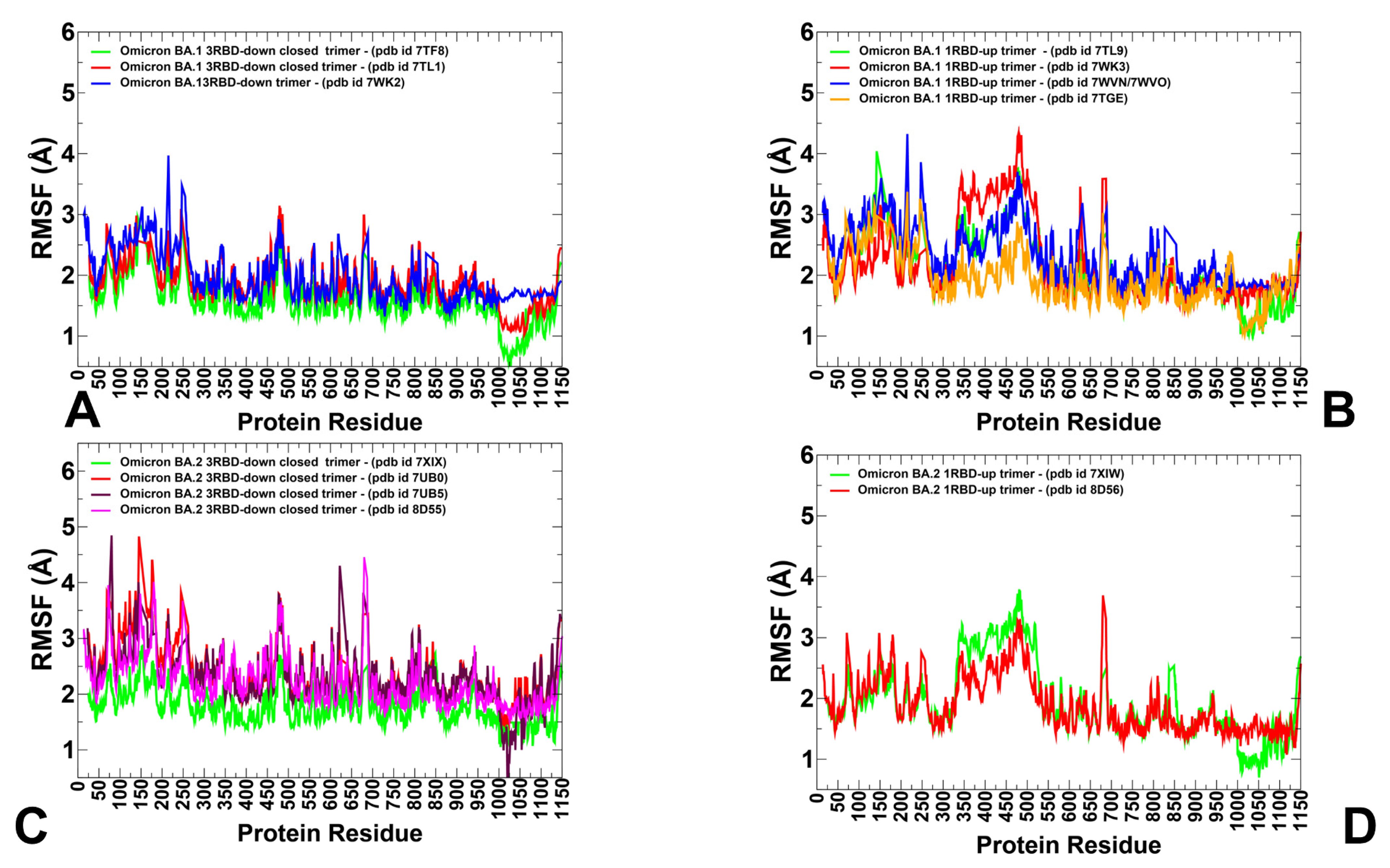
3.1. Conformational Landscapes of Multiple Conformational States of the SARS-CoV-2 S BA.1 and BA.2 Trimers
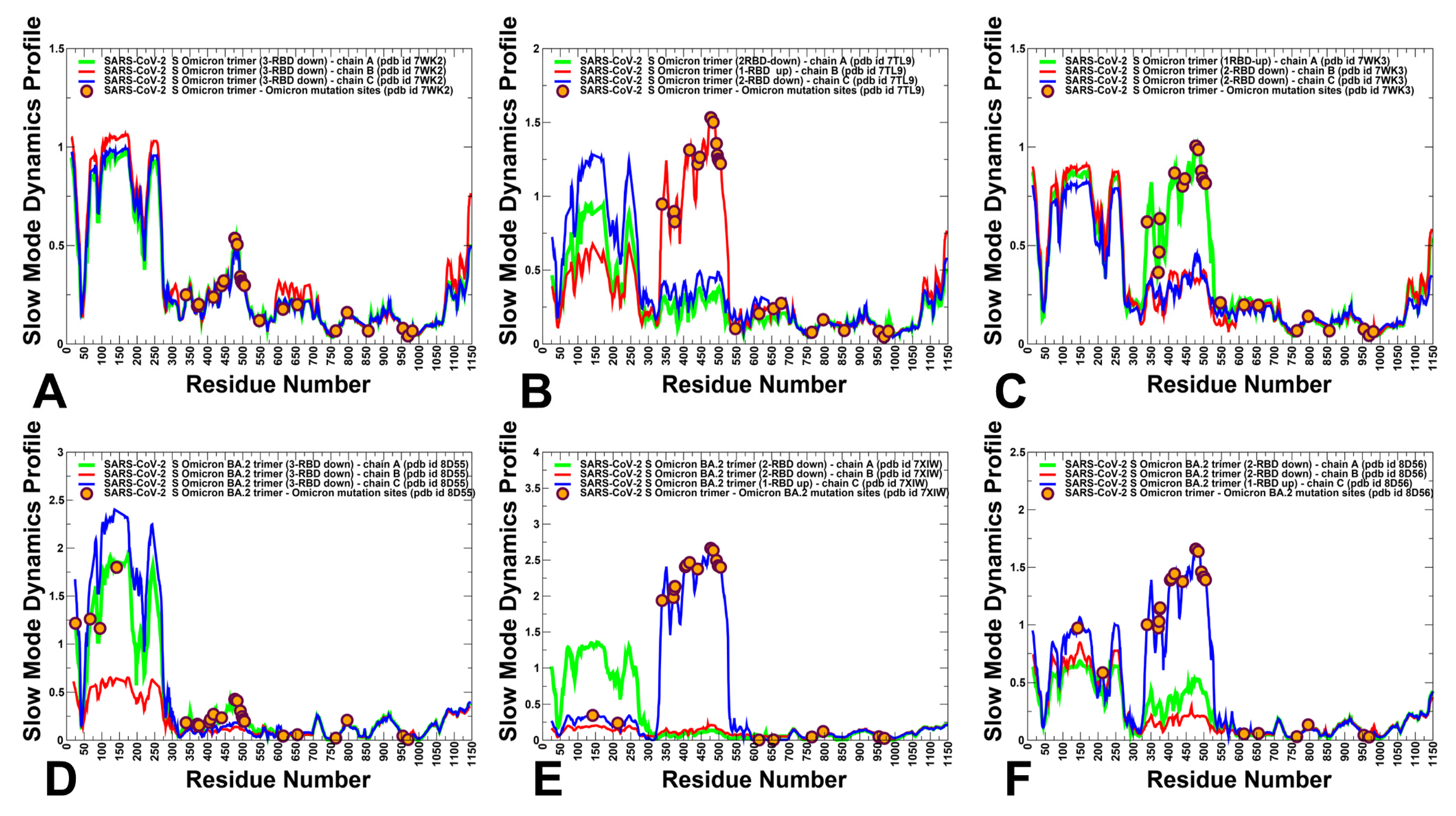
3.2. The Functional Dynamics of the BA.1 and BA.2 Trimers Highlight the Complementary Roles of the Omicron Mutational Sites as Hinges and Transmitters of Collective Motions
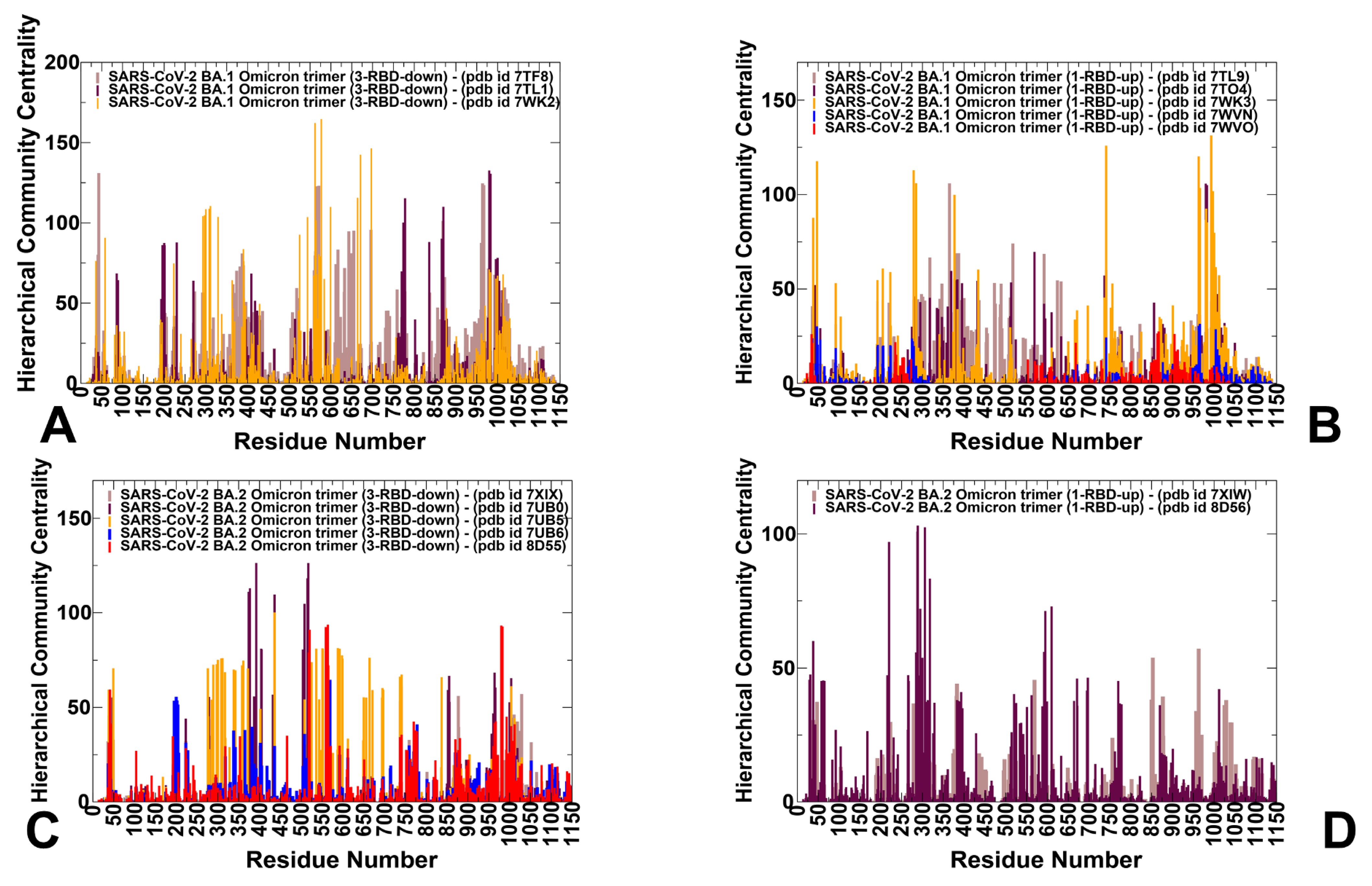
3.3. Dynamic Network Analysis: The Variant-Induced Modulation of Allosteric Mediating Centers
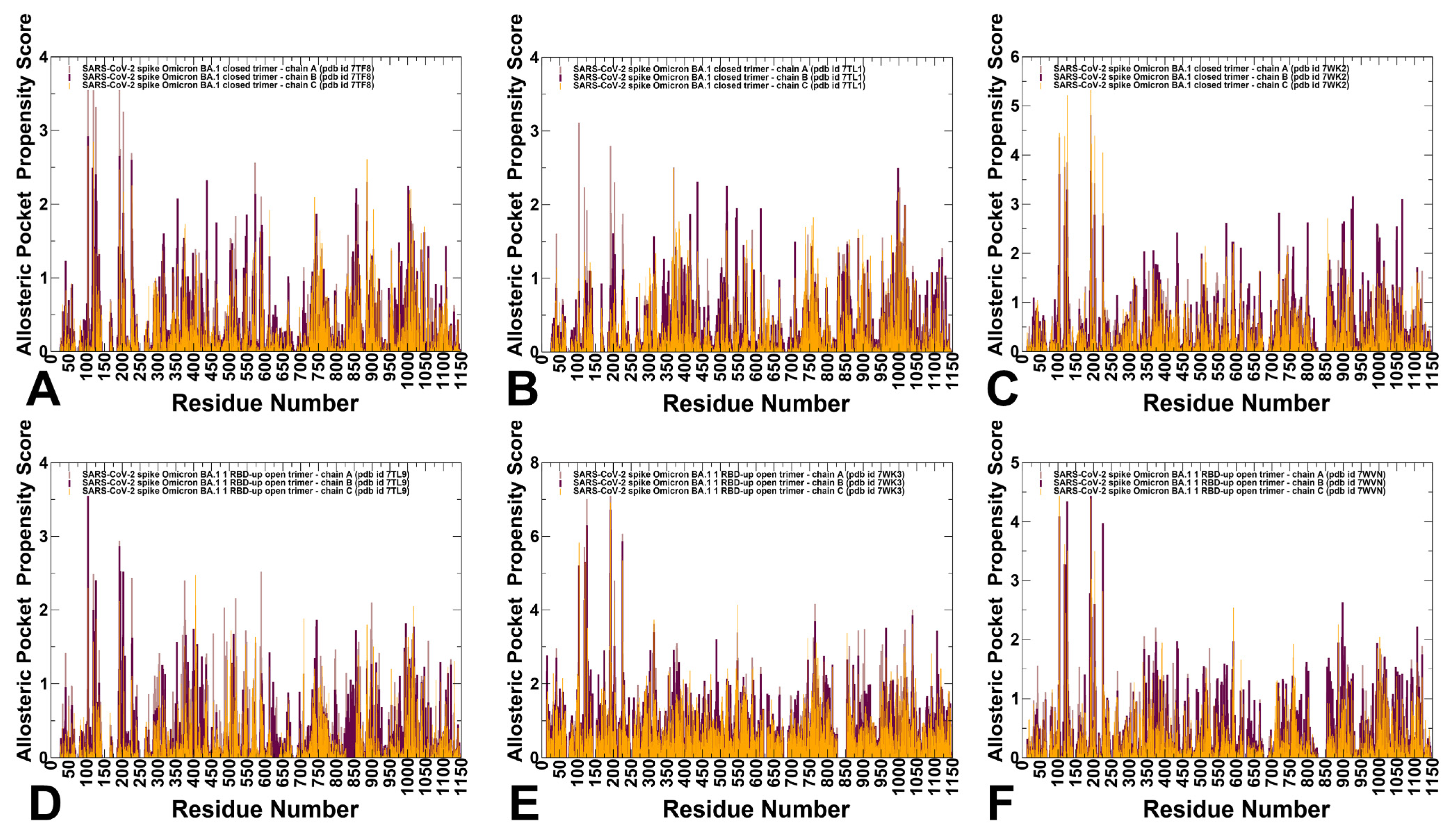
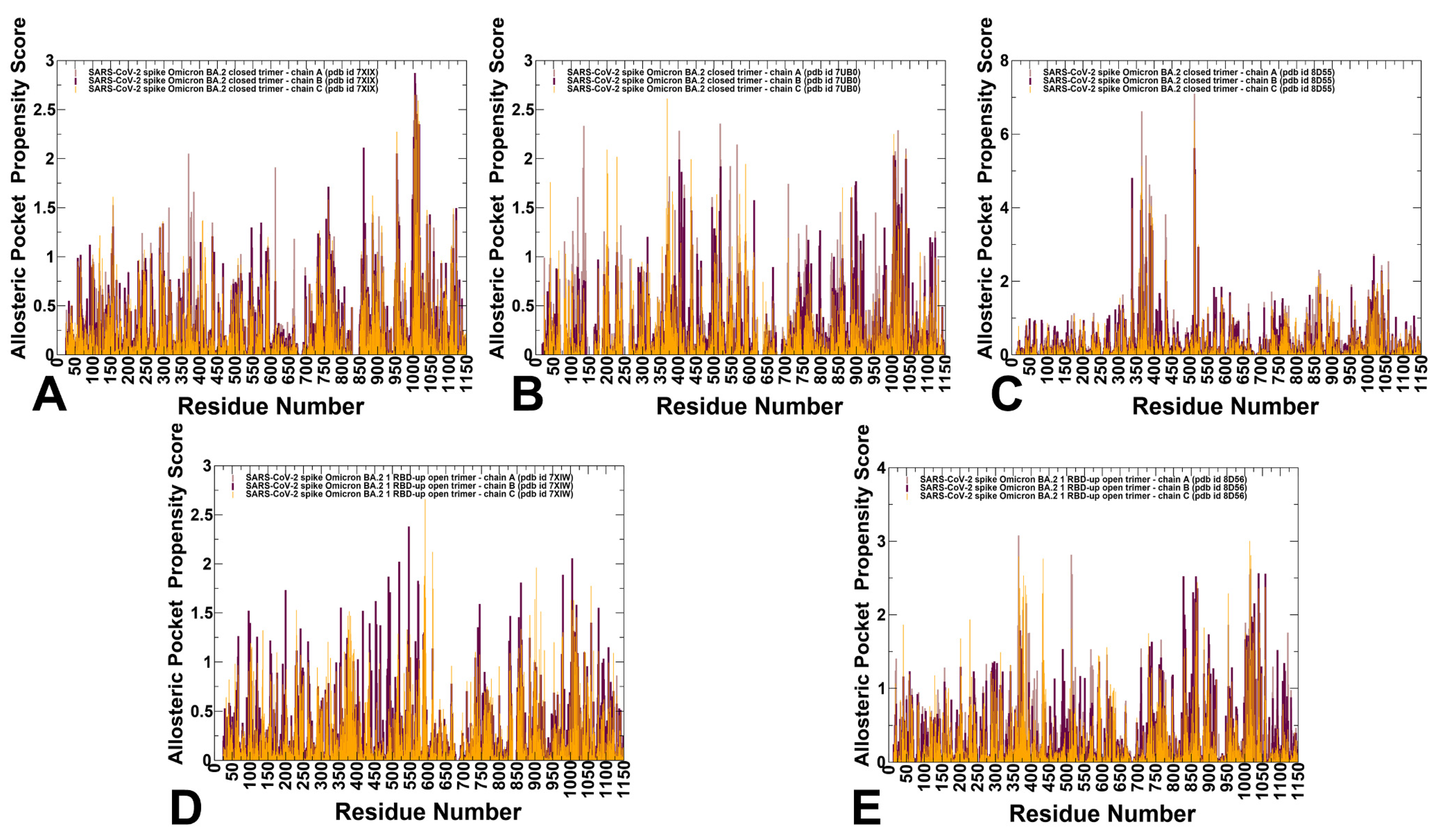
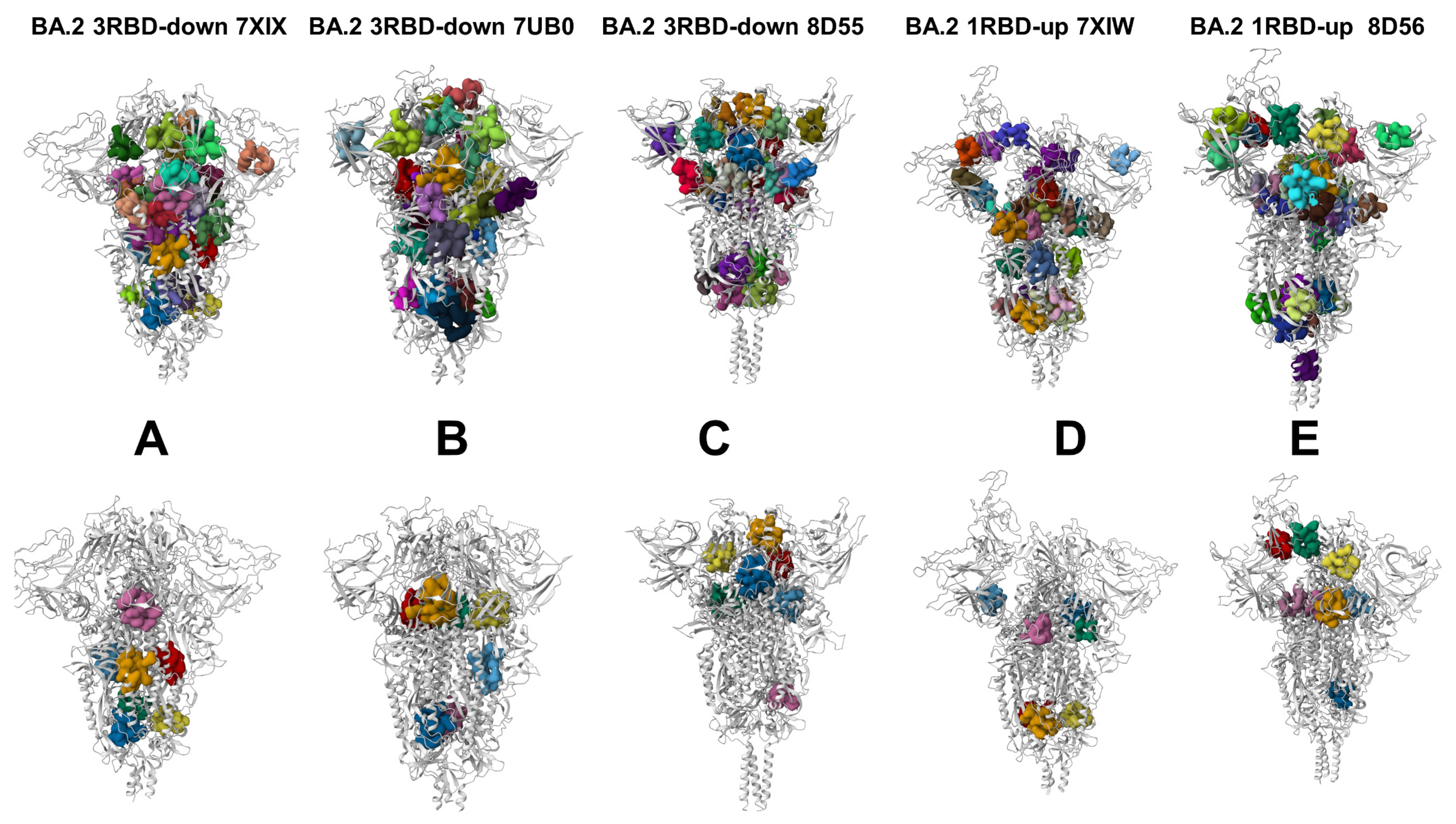
3.4. Allostery-Guided Network Screening of Cryptic Binding Pockets in the S-BA.1 and S-BA.2 Conformational Ensembles: Variant-Specific Modulations of the NTD Binding Sites
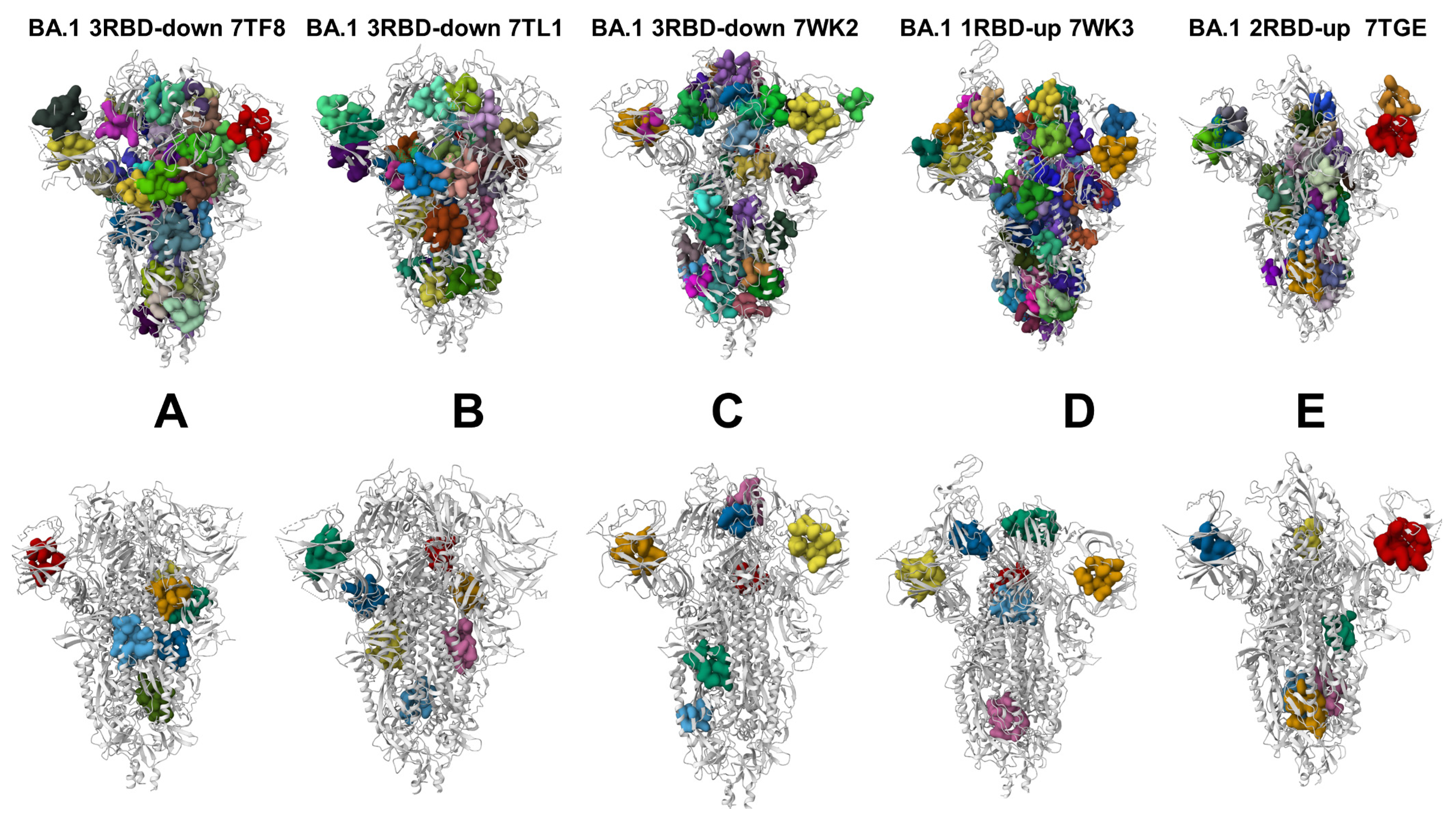
3.5. Deciphering the Anatomy of Cryptic Binding Pockets in the S-BA.1 and S-BA.2 Conformational Ensembles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, A.C.; Mohsin, I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells 2020, 9, 2343. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Carnell, G.W.; Ciazynska, K.A.; Wells, D.A.; Xiong, X.; Aguinam, E.T.; McLaughlin, S.H.; Mallery, D.; Ebrahimi, S.; Ceron-Gutierrez, L.; Asbach, B.; et al. SARS-CoV-2 Spike Protein Stabilized in the Closed State Induces Potent Neutralizing Responses. J. Virol. 2021, 95, e0020321. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, C.; Wang, Y.; Hong, Q.; Zhang, C.; Li, Z.; Xu, S.; Zuo, Q.; Liu, C.; Huang, Z.; et al. Conformational dynamics of the Beta and Kappa SARS-CoV-2 spike proteins and their complexes with ACE2 receptor revealed by cryo-EM. Nat. Commun. 2021, 12, 7345. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In Situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. eLife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Li, X.; Peng, Q.; Su, C.; Huang, B.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular Basis of Receptor Binding and Antibody Neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, L.; Misasi, J.; Pegu, A.; Zhang, Y.; Harris, D.R.; Olia, A.S.; Talana, C.A.; Yang, E.S.; Chen, M.; et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. Science 2022, 376, eabn8897. [Google Scholar] [CrossRef]
- Guo, H.; Gao, Y.; Li, T.; Li, T.; Lu, Y.; Zheng, L.; Liu, Y.; Yang, T.; Luo, F.; Song, S.; et al. Structures of Omicron Spike Complexes and Implications for Neutralizing Antibody Development. Cell Rep. 2022, 39, 110770. [Google Scholar] [CrossRef]
- Stalls, V.; Lindenberger, J.; Gobeil, S.M.-C.; Henderson, R.; Parks, R.; Barr, M.; Deyton, M.; Martin, M.; Janowska, K.; Huang, X.; et al. Cryo-EM Structures of SARS-CoV-2 Omicron BA.2 Spike. Cell Rep. 2022, 39, 111009. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chen, Z.; Zhang, X.; Wen, A.; Yuan, X.; Yu, C.; Yang, J.; He, B.; Cao, Y.; Lu, G. Characterization of SARS-CoV-2 Omicron Spike RBD Reveals Significantly Decreased Stability, Severe Evasion of Neutralizing-Antibody Recognition but Unaffected Engagement by Decoy ACE2 Modified for Enhanced RBD Binding. Signal Transduct. Target. Ther. 2022, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 Mutations Stabilize Spike up-RBD Conformation and Lead to a Non-RBM-Binding Monoclonal Antibody Escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Liu, L.; Liu, L.; Zhang, Z.; Luo, Y.; Huang, Y.; Wang, H.H.; Ho, D.D.; Sheng, Z.; et al. Cryo-EM Structure of the SARS-CoV-2 Omicron Spike. Cell Rep. 2022, 38, 110428. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Liu, B.; Li, F. Cryo-EM Structure of a SARS-CoV-2 Omicron Spike Protein Ectodomain. Nat. Commun. 2022, 13, 1214. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, C.; Cao, X.; Gu, C.; Liu, H.; Jiang, M.; Wang, X.; Yuan, Q.; Wu, K.; Liu, J.; et al. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA.2 variant compared to BA.1 and their possible mouse origins. Cell Res. 2022, 32, 609–620. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and Functional Impact by SARS-CoV-2 Omicron Spike Mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, W.; Gao, H.; Lavine, C.L.; Shi, W.; Peng, H.; Zhu, H.; Anand, K.; Kosikova, M.; Kwon, H.J.; et al. Structural and Functional Characteristics of the SARS-CoV-2 Omicron Subvariant BA.2 Spike Protein. Nat. Struct. Mol. Biol. 2023, 30, 980–990. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.-L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 Spike Reversibly Samples an Open-Trimer Conformation Exposing Novel Epitopes. Nat. Struct. Mol. Biol. 2022, 29, 229–238. [Google Scholar] [CrossRef]
- Calvaresi, V.; Wrobel, A.G.; Toporowska, J.; Hammerschmid, D.; Doores, K.J.; Bradshaw, R.T.; Parsons, R.B.; Benton, D.J.; Roustan, C.; Reading, E.; et al. Structural Dynamics in the Evolution of SARS-CoV-2 Spike Glycoprotein. Nat. Commun. 2023, 14, 1421. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 Neutralizing Antibodies Directed against Spike N-Terminal Domain Target a Single Supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.-W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody Evasion Properties of SARS-CoV-2 Omicron Sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wu, L.; Zheng, A.; Xie, Y.; He, Q.; Rong, X.; Han, P.; Du, P.; Han, P.; Zhang, Z.; et al. Atlas of Currently Available Human Neutralizing Antibodies against SARS-CoV-2 and Escape by Omicron Sub-Variants BA.1/BA.1.1/BA.2/BA.3. Immunity 2022, 55, 1501–1514.e3. [Google Scholar] [CrossRef]
- Marcink, T.C.; Kicmal, T.; Armbruster, E.; Zhang, Z.; Zipursky, G.; Golub, K.L.; Idris, M.; Khao, J.; Drew-Bear, J.; McGill, G.; et al. Intermediates in SARS-CoV-2 Spike–Mediated Cell Entry. Sci. Adv. 2022, 8, eabo3153. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Bowman, G.R.; Juarez-Jimenez, J.; Michel, J.; Gervasio, F.L. Investigating Cryptic Binding Sites by Molecular Dynamics Simulations. Acc. Chem. Res. 2020, 53, 654–661. [Google Scholar] [CrossRef]
- Beglov, D.; Hall, D.R.; Wakefield, A.E.; Luo, L.; Allen, K.N.; Kozakov, D.; Whitty, A.; Vajda, S. Exploring the Structural Origins of Cryptic Sites on Proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3416–E3425. [Google Scholar] [CrossRef]
- Cimermancic, P.; Weinkam, P.; Rettenmaier, T.J.; Bichmann, L.; Keedy, D.A.; Woldeyes, R.A.; Schneidman-Duhovny, D.; Demerdash, O.N.; Mitchell, J.C.; Wells, J.A.; et al. CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites. J. Mol. Biol. 2016, 428, 709–719. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A Highly Conserved Cryptic Epitope in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free Fatty Acid Binding Pocket in the Locked Structure of SARS-CoV-2 Spike Protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Berger, I.; Schaffitzel, C. Cryo-EM Reveals Binding of Linoleic Acid to SARS-CoV-2 Spike Glycoprotein, Suggesting an Antiviral Treatment Strategy. Acta Crystallogr. D Struct. Biol. 2023, 79, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Hodgson, L.; Williamson, M.K.; Buzas, D.; Borucu, U.; Powers, K.; Stenner, R.; Vasileiou, K.; et al. The Free Fatty Acid–Binding Pocket Is a Conserved Hallmark in Pathogenic β-Coronavirus Spike Proteins from SARS-CoV to Omicron. Sci. Adv. 2022, 8, eadc9179. [Google Scholar] [CrossRef] [PubMed]
- Hao, A.; Song, W.; Li, C.; Zhang, X.; Tu, C.; Wang, X.; Wang, P.; Wu, Y.; Ying, T.; Sun, L. Defining a Highly Conserved Cryptic Epitope for Antibody Recognition of SARS-CoV-2 Variants. Signal Transduct. Target. Ther. 2023, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D.; et al. Structural Analysis of Full-Length SARS-CoV-2 Spike Protein from an Advanced Vaccine Candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; dos Santos, M.S.; et al. SARS-CoV-2 Can Recruit a Heme Metabolite to Evade Antibody Immunity. Sci. Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef] [PubMed]
- Altomare, C.G.; Adelsberg, D.C.; Carreno, J.M.; Sapse, I.A.; Amanat, F.; Ellebedy, A.H.; Simon, V.; Krammer, F.; Bajic, G. Structure of a Vaccine-Induced, Germline-Encoded Human Antibody Defines a Neutralizing Epitope on the SARS-CoV-2 Spike N-Terminal Domain. mBio 2022, 13, e0358021. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Juraszek, J.; Rutten, L.; Bakkers, M.J.G.; Blokland, S.; Melchers, J.M.; van den Broek, N.J.F.; Verwilligen, A.Y.W.; Abeywickrema, P.; Vingerhoets, J.; et al. Convergence of Immune Escape Strategies Highlights Plasticity of SARS-CoV-2 Spike. PLoS Pathog. 2023, 19, e1011308. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls the opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 Spike Opening Dynamics and Energetics Reveal the Individual Roles of Glycans and Their Collective Impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of Interactions Regulating Conformational Stability and Dynamics of SARS-CoV-2 S-Protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Dokainish, H.M.; Re, S.; Mori, T.; Kobayashi, C.; Jung, J.; Sugita, Y. The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein. eLife 2022, 11, e75720. [Google Scholar] [CrossRef]
- Brotzakis, Z.F.; Löhr, T.; Vendruscolo, M. Determination of intermediate state structures in the opening pathway of SARS-CoV-2 spike using cryo-electron microscopy. Chem. Sci. 2021, 12, 9168–9175. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Kassab, R.; Krishnan, K. Frustration-driven allosteric regulation and signal transmission in the SARS-CoV-2 spike omicron trimer structures: A crosstalk of the omicron mutation sites allosterically regulates tradeoffs of protein stability and conformational adaptability. Phys. Chem. Chem. Phys. 2022, 24, 17723–17743. [Google Scholar] [CrossRef]
- Zuzic, L.; Samsudin, F.; Shivgan, A.T.; Raghuvamsi, P.V.; Marzinek, J.K.; Boags, A.; Pedebos, C.; Tulsian, N.K.; Warwicker, J.; MacAry, P.; et al. Uncovering Cryptic Pockets in the SARS-CoV-2 Spike Glycoprotein. Structure 2022, 30, 1062–1074.e4. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Kelly, B.; Valant, C.; Michaelis, J.A.; Mastromihalis, O.; Thompson, G.; Venkatakrishnan, A.J.; Hertig, S.; Scammells, P.J.; Sexton, P.M.; et al. Cryptic Pocket Formation Underlies Allosteric Modulator Selectivity at Muscarinic GPCRs. Nat. Commun. 2019, 10, 3289. [Google Scholar] [CrossRef] [PubMed]
- Sztain, T.; Amaro, R.; McCammon, J.A. Elucidation of Cryptic and Allosteric Pockets within the SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2021, 61, 3495–3501. [Google Scholar] [CrossRef] [PubMed]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated Ligand-Binding Site Predictions for Experimental and Modelled Protein Structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef] [PubMed]
- Kozlovskii, I.; Popov, P. Spatiotemporal identification of druggable binding sites using deep learning. Commun. Biol. 2020, 3, 618. [Google Scholar] [CrossRef]
- Xiao, S.; Tian, H.; Tao, P. PASSer2.0: Accurate Prediction of Protein Allosteric Sites Through Automated Machine Learning. Front. Mol. Biosci. 2022, 9, 879251. [Google Scholar] [CrossRef]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSer: Fast and Accurate Prediction of Protein Allosteric Sites. Nucleic Acids Res. 2023, 51, W427–W431. [Google Scholar] [CrossRef]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSerRank: Prediction of Allosteric Sites with Learning to Rank. J. Comput. Chem. 2023, 44, 2223–2229. [Google Scholar] [CrossRef]
- Meller, A.; Bhakat, S.; Solieva, S.; Bowman, G.R. Accelerating Cryptic Pocket Discovery Using AlphaFold. J. Chem. Theory Comput. 2023, 19, 4355–4363. [Google Scholar] [CrossRef]
- Meller, A.; Ward, M.; Borowsky, J.; Kshirsagar, M.; Lotthammer, J.M.; Oviedo, F.; Ferres, J.L.; Bowman, G.R. Predicting Locations of Cryptic Pockets from Single Protein Structures Using the PocketMiner Graph Neural Network. Nat. Commun. 2023, 14, 1177. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.A.; Frederick, T.E.; Mallimadugula, U.L.; Singh, S.; Vithani, N.; Zimmerman, M.I.; Porter, J.R.; Moeder, K.E.; Amarasinghe, G.K.; Bowman, G.R. A Cryptic Pocket in Ebola VP35 Allosterically Controls RNA Binding. Nat. Commun. 2022, 13, 2269. [Google Scholar] [CrossRef]
- Ward, M.D.; Zimmerman, M.I.; Meller, A.; Chung, M.; Swamidass, S.J.; Bowman, G.R. Deep learning the structural determinants of protein biochemical properties by comparing structural ensembles with DiffNets. Nat. Commun. 2021, 12, 3023. [Google Scholar] [CrossRef]
- Meller, A.; Lotthammer, J.M.; Smith, L.G.; Novak, B.; Lee, L.A.; Kuhn, C.C.; Greenberg, L.; Leinwand, L.A.; Greenberg, M.J.; Bowman, G.R. Drug Specificity and Affinity Are Encoded in the Probability of Cryptic Pocket Opening in Myosin Motor Domains. eLife 2023, 12, e83602. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; De Oliveira, S.; Davtyan, A.; Abramyan, T.; Bowman, G.R.; van den Bedem, H. Discovery of a Cryptic Pocket in the AI-Predicted Structure of PPM1D Phosphatase Explains the Binding Site and Potency of Its Allosteric Inhibitors. Front. Mol. Biosci. 2023, 10, 1171143. [Google Scholar] [CrossRef] [PubMed]
- Ghoula, M.; Naceri, S.; Sitruk, S.; Flatters, D.; Moroy, G.; Camproux, A.C. Identifying Promising Druggable Binding Sites and Their Flexibility to Target the Receptor-Binding Domain of SARS-CoV-2 Spike Protein. Comput. Struct. Biotechnol. J. 2023, 21, 2339–2351. [Google Scholar] [CrossRef]
- Davies, S.P.; Mycroft-West, C.J.; Pagani, I.; Hill, H.J.; Chen, Y.-H.; Karlsson, R.; Bagdonaite, I.; Guimond, S.E.; Stamataki, Z.; De Lima, M.A.; et al. The Hyperlipidaemic Drug Fenofibrate Significantly Reduces Infection by SARS-CoV-2 in Cell Culture Models. Front. Pharmacol. 2021, 12, 660490. [Google Scholar] [CrossRef]
- Huang, J.; Chan, K.C.; Zhou, R. Novel Inhibitory Role of Fenofibric Acid by Targeting Cryptic Site on the RBD of SARS-CoV-2. Biomolecules 2023, 13, 359. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Liu, Y.; Kong, R.; Ni, D.; Yu, Z.; Lu, S.; Zhang, J. Harnessing Reversed Allosteric Communication: A Novel Strategy for Allosteric Drug Discovery. J. Med. Chem. 2021, 64, 17728–17743. [Google Scholar] [CrossRef]
- Tee, W.-V.; Guarnera, E.; Berezovsky, I.N. Reversing allosteric communication: From detecting allosteric sites to inducing and tuning targeted allosteric response. PLoS Comput. Biol. 2018, 14, e1006228. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2020, 12, 464–476. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets. Int. J. Mol. Sci. 2022, 23, 11542. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Zhang, Y.; Zhang, X.; Zhang, L.; Shang, W.; Bai, F. Probing the Allosteric Inhibition Mechanism of a Spike Protein Using Molecular Dynamics Simulations and Active Compound Identifications. J. Med. Chem. 2021, 65, 2827–2835. [Google Scholar] [CrossRef]
- Li, B.; Wang, L.; Ge, H.; Zhang, X.; Ren, P.; Guo, Y.; Chen, W.; Li, J.; Zhu, W.; Chen, W.; et al. Identification of Potential Binding Sites of Sialic Acids on the RBD Domain of SARS-CoV-2 Spike Protein. Front. Chem. 2021, 9, 659764. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lin, S.; Chen, Z.; Cao, Y.; He, B.; Lu, G. Targetable Elements in SARS-CoV-2 S2 Subunit for the Design of Pan-Coronavirus Fusion Inhibitors and Vaccines. Signal Transduct. Target. Ther. 2023, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Su, D.; Sun, Y.; Huang, X.; Liang, Y.; Fang, L.; Ma, Y.; Li, W.; Liang, P.; Zheng, S. Cryo-Electron Microscopy Structure of S-Trimer, a Subunit Vaccine Candidate for COVID-19. J. Virol. 2021, 95, e00194-21. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An Interactive Web Server for Efficient Protein Structure Refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Sacquin-Mora, S. Bridging Enzymatic Structure Function via Mechanics: A Coarse-Grain Approach. Methods Enzymol. 2016, 578, 227–248. [Google Scholar] [CrossRef]
- Zacharias, M. Protein–protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003, 12, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Ermak, D.L.; McCammon, J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978, 69, 1352–1360. [Google Scholar] [CrossRef]
- Pastor, R.W.; Venable, R.; Karplus, M. Brownian dynamics simulation of a lipid chain in a membrane bilayer. J. Chem. Phys. 1988, 89, 1112–1127. [Google Scholar] [CrossRef]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef]
- Lombardi, L.E.; Marti, M.A.; Capece, L. CG2AA: Backmapping protein coarse-grained structures. Bioinformatics 2016, 32, 1235–1237. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: A community-hopping model of allosteric regulation and communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: Fast Generation of Probabilistic Residue Interaction Networks from Structural Ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; Scientific Research: Atlanta, GA, USA, 2011; pp. 11–15. [Google Scholar]
- Girvan, M.; Newman, M.E. Community Structure in Social and Biological Networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Astl, L.; Verkhivker, G.M. Atomistic Modeling of the ABL Kinase Regulation by Allosteric Modulators Using Structural Perturbation Analysis and Community-Based Network Reconstruction of Allosteric Communications. J. Chem. Theory Comput. 2019, 15, 3362–3380. [Google Scholar] [CrossRef]
- Jensen, P.; Morini, M.; Karsai, M.; Venturini, T.; Vespignani, A.; Jacomy, M.; Cointet, J.-P.; Mercklé, P.; Fleury, E. Detecting Global Bridges in Networks. J. Complex Netw. 2016, 4, 319–329. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. In Advances in Neural Information Processing Systems, Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; The MIT Press: Cambridge, MA, USA, 2017; Volume 30. [Google Scholar]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Lavery, R.; Sacquin-Mora, S. Protein Mechanics: A Route from Structure to Function. J. Biosci. 2007, 32, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Sacquin-Mora, S. Motions and Mechanics: Investigating Conformational Transitions in Multi-Domain Proteins with Coarse-Grain Simulations. Mol. Simul. 2013, 40, 229–236. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef]
- Palma, J.; Pierdominici-Sottile, G. On the Uses of PCA to Characterise Molecular Dynamics Simulations of Biological Macromolecules: Basics and Tips for an Effective Use. ChemPhysChem 2023, 24, e202200491. [Google Scholar] [CrossRef]
- Braet, S.M.; Buckley, T.S.; Venkatakrishnan, V.; Dam, K.-M.A.; Bjorkman, P.J.; Anand, G.S. Timeline of Changes in Spike Conformational Dynamics in Emergent SARS-CoV-2 Variants Reveal Progressive Stabilization of Trimer Stalk with Altered NTD Dynamics. eLife 2023, 12, e82584. [Google Scholar] [CrossRef]
- Raghuvamsi, P.V.; Tulsian, N.K.; Samsudin, F.; Qian, X.; Purushotorman, K.; Yue, G.; Kozma, M.M.; Hwa, W.Y.; Lescar, J.; Bond, P.J.; et al. SARS-CoV-2 S Protein:ACE2 Interaction Reveals Novel Allosteric Targets. eLife 2021, 10, e63646. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, R.; Hodge, E.A.; Díaz-Salinas, M.A.; Nguyen, A.; Munro, J.B.; Lee, K.K. hACE2-Induced Allosteric Activation in SARS-CoV versus SARS-CoV-2 Spike Assemblies Revealed by Structural Dynamics. ACS Infect. Dis. 2023, 9, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-Terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Andreano, E.; Paciello, I.; Marchese, S.; Donnici, L.; Pierleoni, G.; Piccini, G.; Manganaro, N.; Pantano, E.; Abbiento, V.; Pileri, P.; et al. Anatomy of Omicron BA.1 and BA.2 Neutralizing Antibodies in COVID-19 mRNA Vaccinees. Nat. Commun. 2022, 13, 3375. [Google Scholar] [CrossRef]
- Shoemark, D.K.; Oliveira, A.S.F.; Davidson, A.D.; Berger, I.; Schaffitzel, C.; Mulholland, A.J. Molecular Dynamics of Spike Variants in the Locked Conformation: RBD Interfaces, Fatty Acid Binding and Furin Cleavage Sites. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The Anti-Influenza Virus Drug, Arbidol Is an Efficient Inhibitor of SARS-CoV-2 in Vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A Potential Antiviral Drug for the Treatment of SARS-CoV-2 by Blocking Trimerization of the Spike Glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Silva, R.P.; Huang, Y.; Nguyen, A.W.; Hsieh, C.-L.; Olaluwoye, O.S.; Kaoud, T.S.; Wilen, R.E.; Qerqez, A.N.; Park, J.-G.; Khalil, A.M.; et al. Identification of a Conserved S2 Epitope Present on Spike Proteins from All Highly Pathogenic Coronaviruses. eLife 2023, 12, e83710. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omicron Variant | Mutational Landscape |
|---|---|
| BA.1 | A67, T95I, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, and L981F |
| BA.2 | T19I, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, and N969K |
| Variant | PDB Code | Description | RBD Orientation | Resolution | Ref. |
|---|---|---|---|---|---|
| BA.1 | 7TF8 | S Omicron Trimer, closed | 3 RBD-down | 3.9 | [27] |
| BA.1 | 7WK2 | S Omicron Trimer, closed | 3 RBD-down | 3.1 | [24] |
| BA.1 | 7TNW | S Omicron Trimer, closed | 3 RBD-down | 3.1 | [37] |
| BA.1 | 7TL1 | S Omicron Trimer, closed | 3 RBD-down | 3.5 | [27] |
| BA.1 | 7TL9 | S Omicron Trimer, open | 1 RBD-up | 3.5 | [27] |
| BA.1 | 7TEI | S Omicron Trimer, open | 1 RBD-up | 3.4 | [27] |
| BA.1 | 7WK3 | S Omicron Trimer, open | 1 RBD-up | 3.4 | [24] |
| BA.1 | 7TO4 | S Omicron Trimer, open | 1 RBD-up | 3.4 | [37] |
| BA.1 | 7WVN | S Omicron Trimer, open | 1 RBD-up | 4.0 | [24] |
| BA.1 | 7WVO | S Omicron Trimer, open | 1 RBD-up | 4.0 | [24] |
| BA.1 | 7TGE | S Omicron Trimer, open | 2 RBD-up | 3.7 | [27] |
| BA.2 | 7XIX | S Omicron Trimer, closed | 3 RBD-down | 3.25 | [38] |
| BA.2 | 7UB0 | S Omicron Trimer, closed | 3 RBD-down | 3.31 | [31] |
| BA.2 | 7UB5 | S Omicron Trimer, closed | 3 RBD-down | 3.35 | [31] |
| BA.2 | 7UB6 | S Omicron Trimer, closed | 3 RBD-down | 3.52 | [31] |
| BA.2 | 8D55 | S Omicron Trimer, closed | 3 RBD-down | 2.8 | [37] |
| BA.2 | 7XIW | S Omicron Trimer, open | 1 RBD-up | 3.62 | [38] |
| BA.2 | 8D56 | S Omicron Trimer, open | 1 RBD-up | 3.0 | [37] |
| PDB | System | CG-BD | # Simulations | All-Atom MD | |
|---|---|---|---|---|---|
| 7WK2 | S-BA.1 | 3-RBD down | 500,000 steps | 100 | 500 ns |
| 7WK3 | S-BA.1 | 1-RBD up | 500,000 steps | 100 | 500 ns |
| 7XIX | S-BA.2 | 3-RBD down | 500,000 steps | 100 | 500 ns |
| 7XIW | S-BA.2 | 1-RBD up | 500,000 steps | 100 | 500 ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G.; Alshahrani, M.; Gupta, G. Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites. Viruses 2023, 15, 2009. https://doi.org/10.3390/v15102009
Verkhivker G, Alshahrani M, Gupta G. Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites. Viruses. 2023; 15(10):2009. https://doi.org/10.3390/v15102009
Chicago/Turabian StyleVerkhivker, Gennady, Mohammed Alshahrani, and Grace Gupta. 2023. "Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites" Viruses 15, no. 10: 2009. https://doi.org/10.3390/v15102009
APA StyleVerkhivker, G., Alshahrani, M., & Gupta, G. (2023). Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites. Viruses, 15(10), 2009. https://doi.org/10.3390/v15102009