

Establishment of an In Vitro Model of Pseudorabies Virus Latency and Reactivation and Identification of Key Viral Latency-Associated Genes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. One-Step and Multistep Growth Curves and Plaque Assay
2.3. PCR, qPCR, and RT-qPCR
2.4. Transmission Electronic Microscopy
2.5. Western Blotting
2.6. Thermoregulation Model of Latent Infection and Reactivation of PRV
2.7. Statistical Analysis
3. Results
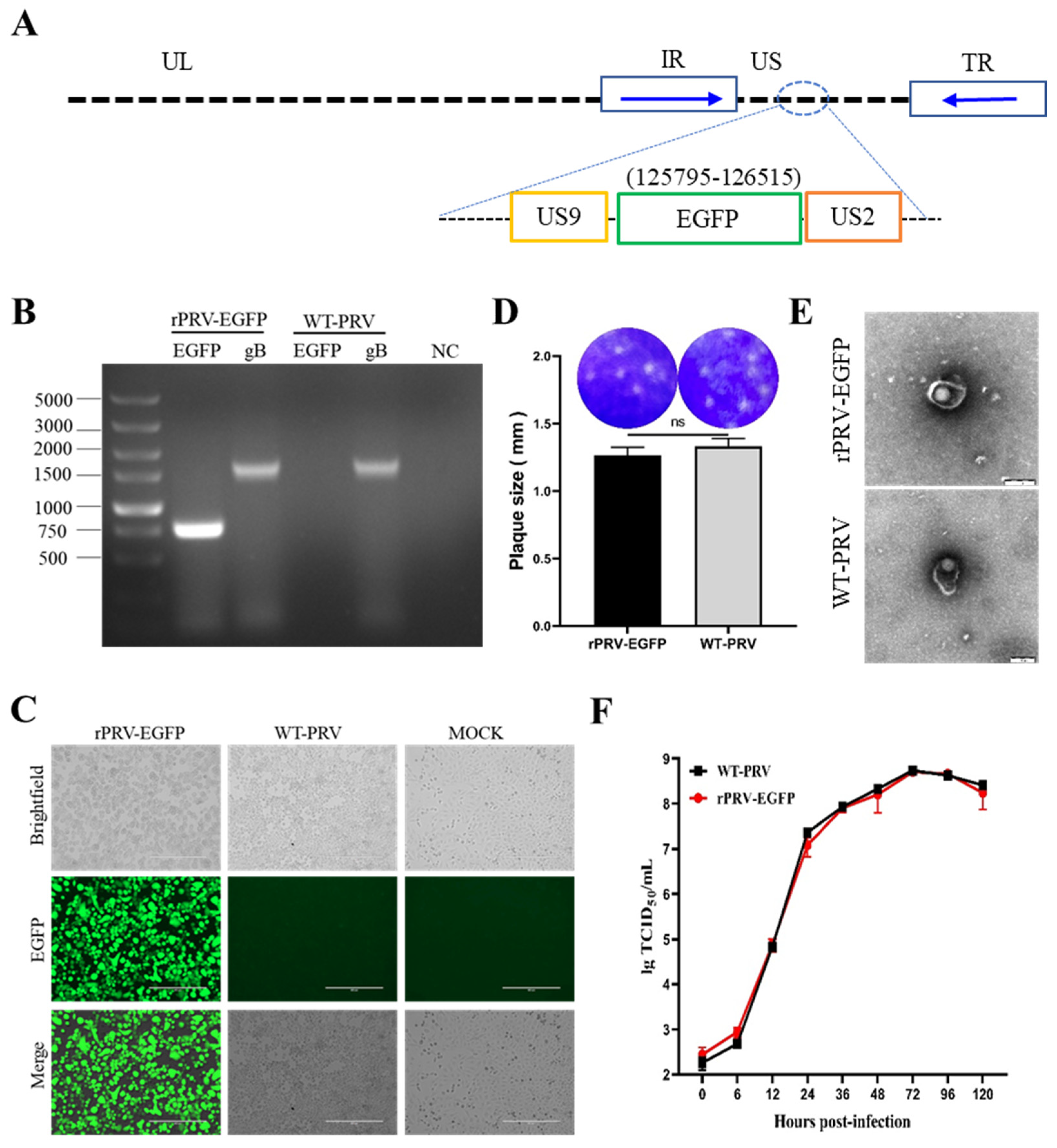
3.1. Generation and Identification of rPRV-EGFP
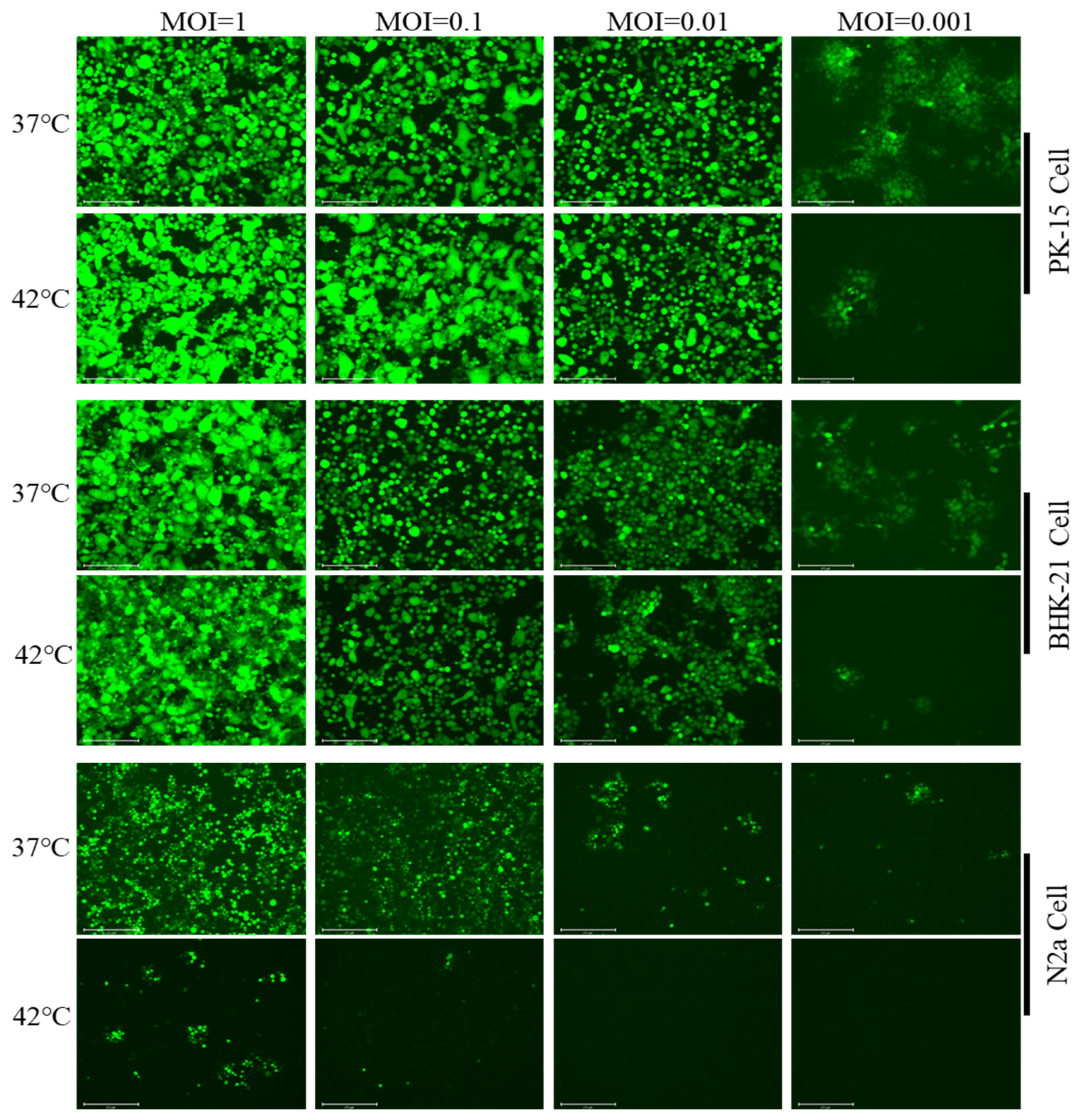
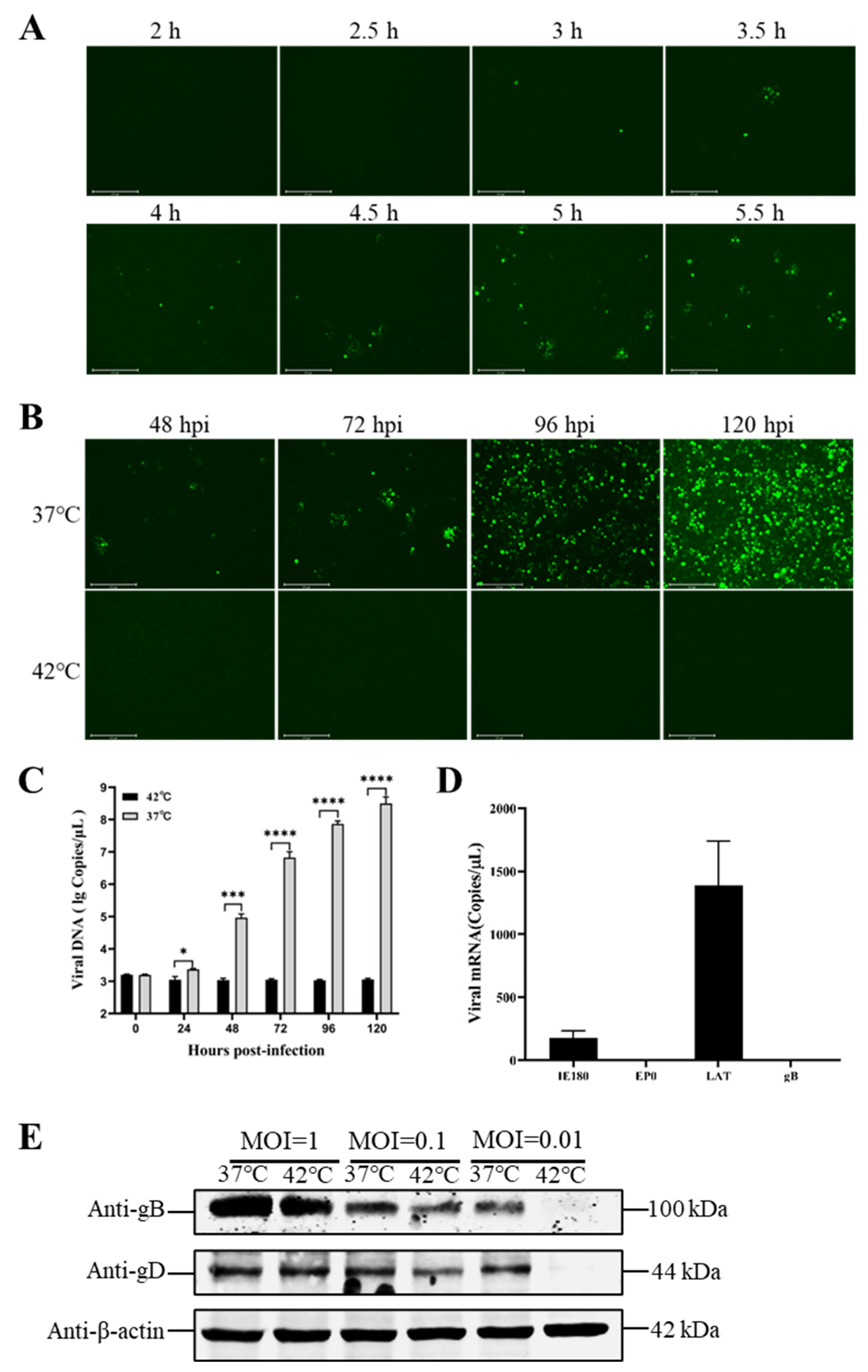
3.2. Hyperthermic Stress Maintained PRV Latency in Neuron-like Cells
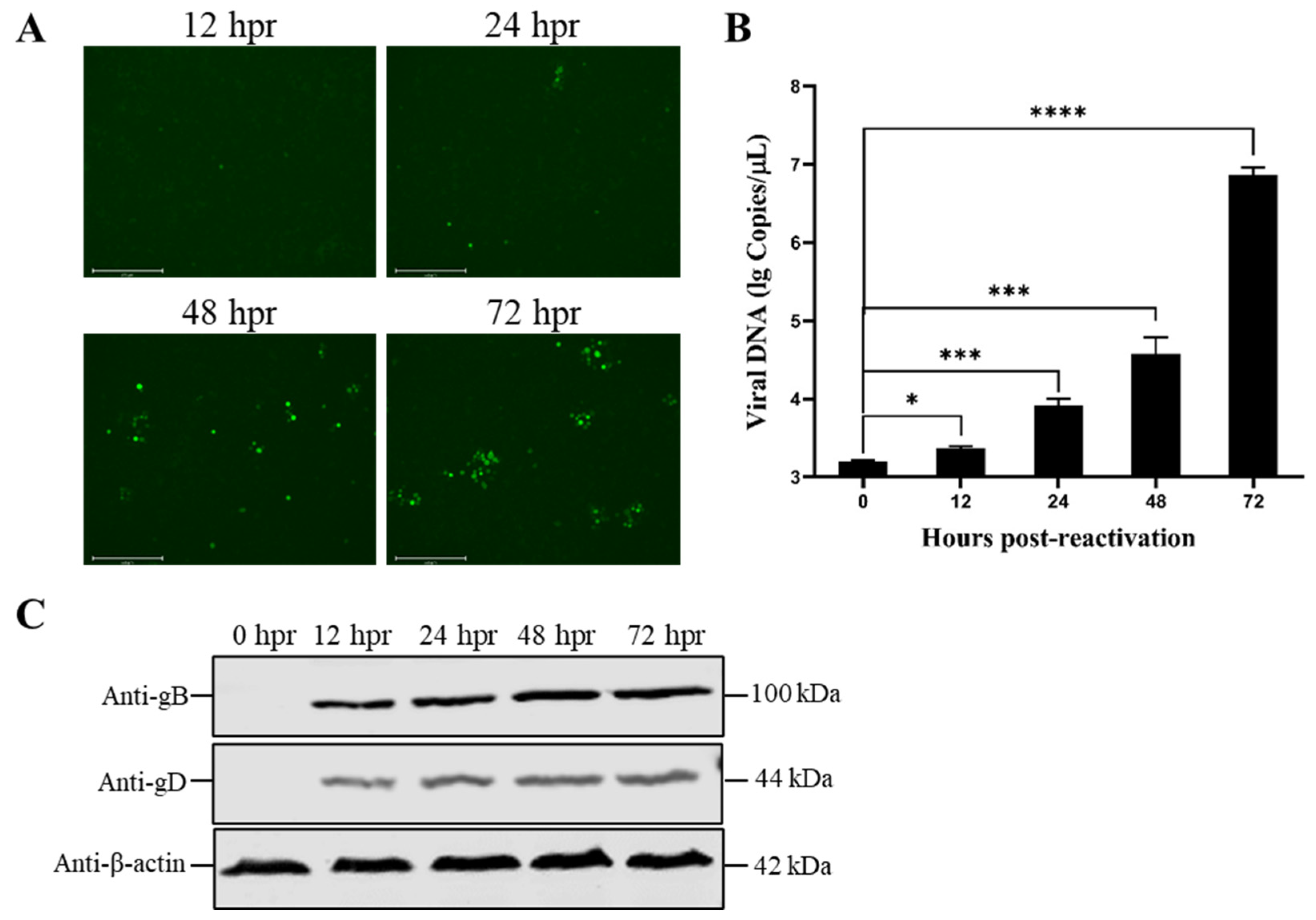
3.3. Reduced Temperature Enhanced the Reactivation of the Latent PRV
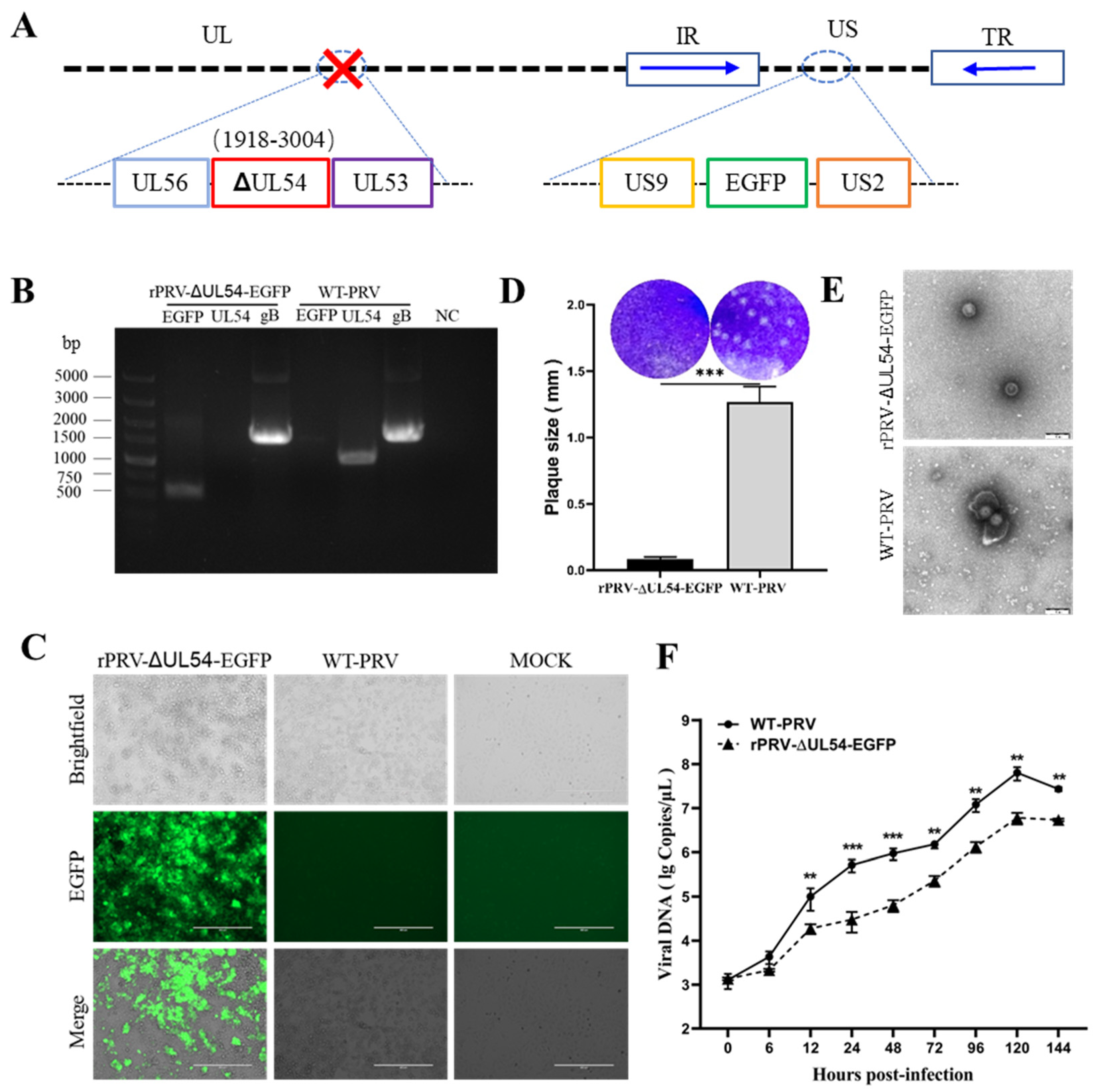
3.4. Deleting the UL54 Gene Affects Viral Particle Morphology and Replication Efficiency
3.5. Deleting the UL54 Gene Affects PRV Reactivation without Affecting Latency
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Laval, K.; Vernejoul, J.B.; Van Cleemput, J.; Koyuncu, O.O.; Enquist, L.W. Virulent Pseudorabies Virus Infection Induces a Specific and Lethal Systemic Inflammatory Response in Mice. J. Virol. 2018, 92, 24. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, H.; Qi, H.; Li, H.; Pan, L.; Li, L.F.; Zhang, K.; Yuan, M.; Wang, Y.; Qiu, H.J.; et al. Histamine is Responsible for the Neuropathic Itch Induced by the Pseudorabies Virus Variant in a Mouse Model. Viruses 2022, 14, 1067. [Google Scholar] [CrossRef]
- Bailer, S.M. Venture from the Interior-Herpesvirus pUL31 Escorts Capsids from Nucleoplasmic Replication Compartments to Sites of Primary Envelopment at the Inner Nuclear Membrane. Cells 2017, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.C.; Mohr, I. A Cultured Affair: HSV Latency and Reactivation in Neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Bloom, D.C. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv. Virus Res. 2016, 94, 53–80. [Google Scholar] [PubMed]
- Lu, J.; Yuan, W.; Zhu, Y.; Hou, S.; Wang, X. Latent Pseudorabies Virus Infection in Medulla Oblongata from Quarantined Pigs. Transbound. Emerg. Dis. 2021, 68, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.; Oxman, M.N.; et al. Varicella Zoster Virus Infection. Nat. Rev. Dis. Prim. 2015, 1, 15016. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Beer, T.; Cohrs, R.; Gilden, D.H. Configuration of Latent Varicella-Zoster Virus-DNA. J. Virol. 1995, 69, 8151–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G. Herpesvirus Transport to the Nervous System and Back Again. Ann. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef] [Green Version]
- Reese, T.A. Coinfections: Another Variable in the Herpesvirus Latency-Reactivation Dynamic. J. Virol. 2016, 90, 5534–5537. [Google Scholar] [CrossRef] [Green Version]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived Neurons. PLoS Pathog. 2015, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.; Zhu, S.; Pearce, C.M.; Viejo-Borbolla, A. Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses 2019, 11, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Srinivas, K.; Mohr, I.; Huang, T.; Wilson, A. Using Primary SCG Neuron Cultures to Study Molecular Determinants of HSV-1 Latency and Reactivation. Methods Mol. Biol. 2020, 2060, 263–277. [Google Scholar]
- Huang, Y.; Chien, M.; Wu, C.; Huang, C. Mapping of Functional Regions Conferring Nuclear Localization and RNA-Binding Activity of Pseudorabies Virus Early Protein UL54. J. Virol. Methods 2005, 130, 102–107. [Google Scholar] [CrossRef]
- Li, M.; Wang, S.; Cai, M.; Zheng, C. Identification of Nuclear and Nucleolar Localization Signals of Pseudorabies Virus (PRV) Early Protein UL54 Reveals that Its Nuclear Targeting Is Required for Efficient Production of PRV. J. Virol. 2011, 85, 10239–10251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostler, J.B.; Jones, C. Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses 2021, 13, 2296. [Google Scholar] [CrossRef]
- Luo, Y.; Li, N.; Cong, X.; Wang, C.H.; Du, M.; Li, L.; Zhao, B.; Yuan, J.; Liu, D.D.; Li, S.; et al. Pathogenicity and Genomic Characterization of a Pseudorabies Virus Variant Isolated from Bartha-K61-Vaccinated Swine Population in China. Vet. Microbiol. 2014, 174, 107–115. [Google Scholar] [CrossRef]
- Zhou, M.; Abid, M.; Yin, H.; Wu, H.; Teklue, T.; Qiu, H.J.; Sun, Y. Establishment of An Efficient and Flexible Genetic Manipulation Platform Based on a Fosmid Library for Rapid Generation of Recombinant Pseudorabies Virus. Front. Microbiol. 2018, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.; Teklue, T.; Li, Y.F.; Wu, H.; Wang, T.; Qiu, H.J.; Sun, Y. Generation and Immunogenicity of a Recombinant Pseudorabies Virus Co-Expressing Classical Swine Fever Virus E2 Protein and Porcine Circovirus Type 2 Capsid Protein Based on Fosmid Library Platform. Pathogens 2019, 8, 279. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Wu, H.; Abid, M.; Qiu, H.J.; Sun, Y. Establishment of a Fosmid Library for Pseudorabies Virus SC Strain and Application in Viral Neuronal Tracing. Front. Microbiol. 2020, 11, 1168. [Google Scholar] [CrossRef]
- Kinchington, P.R.; Bookey, D.; Turse, S.E. The Transcriptional Regulatory Proteins Encoded by Varicella-Zoster Virus Open Reading Frames (Orfs) 4 and 63, But not Orf-61, Are Associated with Purified Virus-Particles. J. Virol. 1995, 69, 4274–4282. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, K.M.; Tank, D.W.; Enquist, L.W. Pseudorabies Virus Infection Alters Neuronal Activity and Connectivity In Vitro. PLoS Pathog. 2009, 5, 10. [Google Scholar] [CrossRef]
- Xiao, K.; Xiong, D.; Chen, G.; Yu, J.; Li, Y.; Chen, K.; Zhang, L.; Xu, Y.; Xu, Q.; Huang, X.; et al. RUNX1-Mediated Alphaherpesvirus-Host Trans-Species Chromatin Interaction Promotes Viral Transcription. Sci. Adv. 2021, 7, 26. [Google Scholar] [CrossRef]
- Krishnan, R.; Stuart, P.M. Developments in Vaccination for Herpes Simplex Virus. Front. Microbiol. 2021, 12, 798927. [Google Scholar] [CrossRef]
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.F.; He, R.R. Disturbed Yin-Yang Balance: Stress Increases the Susceptibility to Primary and Recurrent Infections of Herpes Simplex Virus Type 1. Acta Pharm. Sin. B 2020, 10, 383–398. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus Latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- St Leger, A.J.; Koelle, D.M.; Kinchington, P.R.; Verjans, G. Local Immune Control of Latent Herpes Simplex Virus Type 1 in Ganglia of Mice and Man. Front. Immunol. 2021, 12, 723809. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; MacGibeny, M.A.; Enquist, L.W. Latent Versus Productive Infection: The Alpha Herpesvirus Switch. Future Virol. 2018, 13, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; de Miranda, P.; Beauchamp, L.; Schaeffer, H.J. Selectivity of Action of an Antiherpetic Agent, 9-(2-Hydroxyethoxymethyl) Guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, J.E.; Spector, T. Herpes Simplex Virus Type 1 DNA Polymerase. Mechanism of Inhibition by Acyclovir Triphosphate. J. Biol. Chem. 1989, 264, 7405–7411. [Google Scholar] [CrossRef]
- Mackowiak, P.A. Concepts of Fever. Arch. Intern. Med. 1998, 158, 1870–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.Y.; Wu, X.M.; Che, Y.L.; Chen, R.J.; Hou, B.; Wang, C.Y.; Wang, L.B.; Zhou, L.J. The Immune Efficacy of Inactivated Pseudorabies Vaccine Prepared from FJ-2012ΔgE/gI Strain. Microorganisms 2022, 10, 1880. [Google Scholar] [CrossRef] [PubMed]
- Rafael, D.; Tomer, E.; Kobiler, O. A Single Herpes Simplex Virus 1 Genome Reactivates from Individual Cells. Microbiol. Spectr. 2022, 31, e0114422. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Preston, C.M. Establishment of Latency In Vitro by the Herpes Simplex Virus Type 1 Mutant in1814. J. Gen. Virol. 1991, 72, 907–913. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences (5′ to 3′) |
|---|---|
| US9-rpsl-F | AGAGCTGGTTTAGTGAACCGTCAGATCCGCTAGCGCTACCGGTCGCCACCGGCCTGGTGATGATGGCGGGATC |
| US9-rpsl-R | GGGCGCGGCGGATGGGGGCGGGCCCCCGCTCCCGTTCGCTCGCTCGCTCGTCAGAAGAACTCGTCAAGAAGGC |
| US9-EGFP-F | AGAGCTGGTTTAGTGAACCGTCAGATCCGCTAGCGCTACCGGTCGCCACCCGCTAGCGCTACCGGTCGCCAC |
| US9-EGFP-R | GGGCGCGGCGGATGGGGGCGGGCCCCCGCTCCCGTTCGCTCGCTCGCTCGATAACTTCGTATAGCATACATT |
| US9-JC-F | CGTATTAGTCATCGCTATTAC |
| US9-JC-R | AACAGAGACGCGGAGGAGAGG |
| UL54-rpsl-F | GGTTGCAGTAAAAGTACTTCCCGTGCATGTACACGGGGACGAGGGTGTAGGGCCTGGTGATGATGGCGGGATC |
| UL54-rpsl-R | AACAGCAGCGGCAGCGAGGCGTCCCGGTCGGGGAGCGAGGAGCGGCGCCCTCAGAAGAACTCGTCAAGAAGGC |
| UL54-del-F | GGTCTTGTGGGCGTGAGCCGCGCCCGGACGGGCGGC |
| UL54-del-R | TCCGGGCGCGGCTCACGCCCACAAGACCGGCTGCGA |
| UL54-JC-F | CTCGCGCACGCCAGAGAGGTAC |
| UL54-JC-R | CGCTCGCACCACGGTCATGGAG |
| Primers | Sequences (5′ to 3′) |
|---|---|
| EGFP-JC-F(PCR) | ATGGTGAGCAAGGGCGAGGAG |
| EGFP-JC-R(PCR) | TTACTTGTACAGCTCGTCCAT |
| gB-JC-F(PCR) | ATGGACATGTACCGGATCATGT |
| gB-JC-R(PCR) | AGAGCGTGACGCGCAACTTTCTGC |
| IE180-F(qPCR/RT-qPCR) | CTGGCAGAACTGGTTGAAGC |
| IE180-R(qPCR/RT-qPCR) | TCGTGCGCCTCATCTACAG |
| EP0-F(qPCR/RT-qPCR) | TGCGCCGATATGTCAAACAG |
| EP0-R(qPCR/RT-qPCR) | TCGTGGACAACATCGTCGAG |
| gB-F(qPCR/RT-qPCR) | TCCTCGACGATGCAGTTGAC |
| gB-R(qPCR/RT-qPCR) | ACCAACGACACCTACACCAAG |
| LAT-F(qPCR/RT-qPCR) | ACGTGACGTTTTTGCCGATG |
| LAT-R(qPCR/RT-qPCR) | GCGCGATATGCAGATGAGATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.; Li, M.; Zhang, X.; Xia, Y.; Mian, A.M.; Wu, H.; Sun, Y.; Qiu, H.-J. Establishment of an In Vitro Model of Pseudorabies Virus Latency and Reactivation and Identification of Key Viral Latency-Associated Genes. Viruses 2023, 15, 808. https://doi.org/10.3390/v15030808
Pan L, Li M, Zhang X, Xia Y, Mian AM, Wu H, Sun Y, Qiu H-J. Establishment of an In Vitro Model of Pseudorabies Virus Latency and Reactivation and Identification of Key Viral Latency-Associated Genes. Viruses. 2023; 15(3):808. https://doi.org/10.3390/v15030808
Chicago/Turabian StylePan, Li, Mingzhi Li, Xinyu Zhang, Yu Xia, Assad Moon Mian, Hongxia Wu, Yuan Sun, and Hua-Ji Qiu. 2023. "Establishment of an In Vitro Model of Pseudorabies Virus Latency and Reactivation and Identification of Key Viral Latency-Associated Genes" Viruses 15, no. 3: 808. https://doi.org/10.3390/v15030808
APA StylePan, L., Li, M., Zhang, X., Xia, Y., Mian, A. M., Wu, H., Sun, Y., & Qiu, H. -J. (2023). Establishment of an In Vitro Model of Pseudorabies Virus Latency and Reactivation and Identification of Key Viral Latency-Associated Genes. Viruses, 15(3), 808. https://doi.org/10.3390/v15030808