
Infectivity-Enhanced, Conditionally Replicative Adenovirus for COX-2-Expressing Castration-Resistant Prostate Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
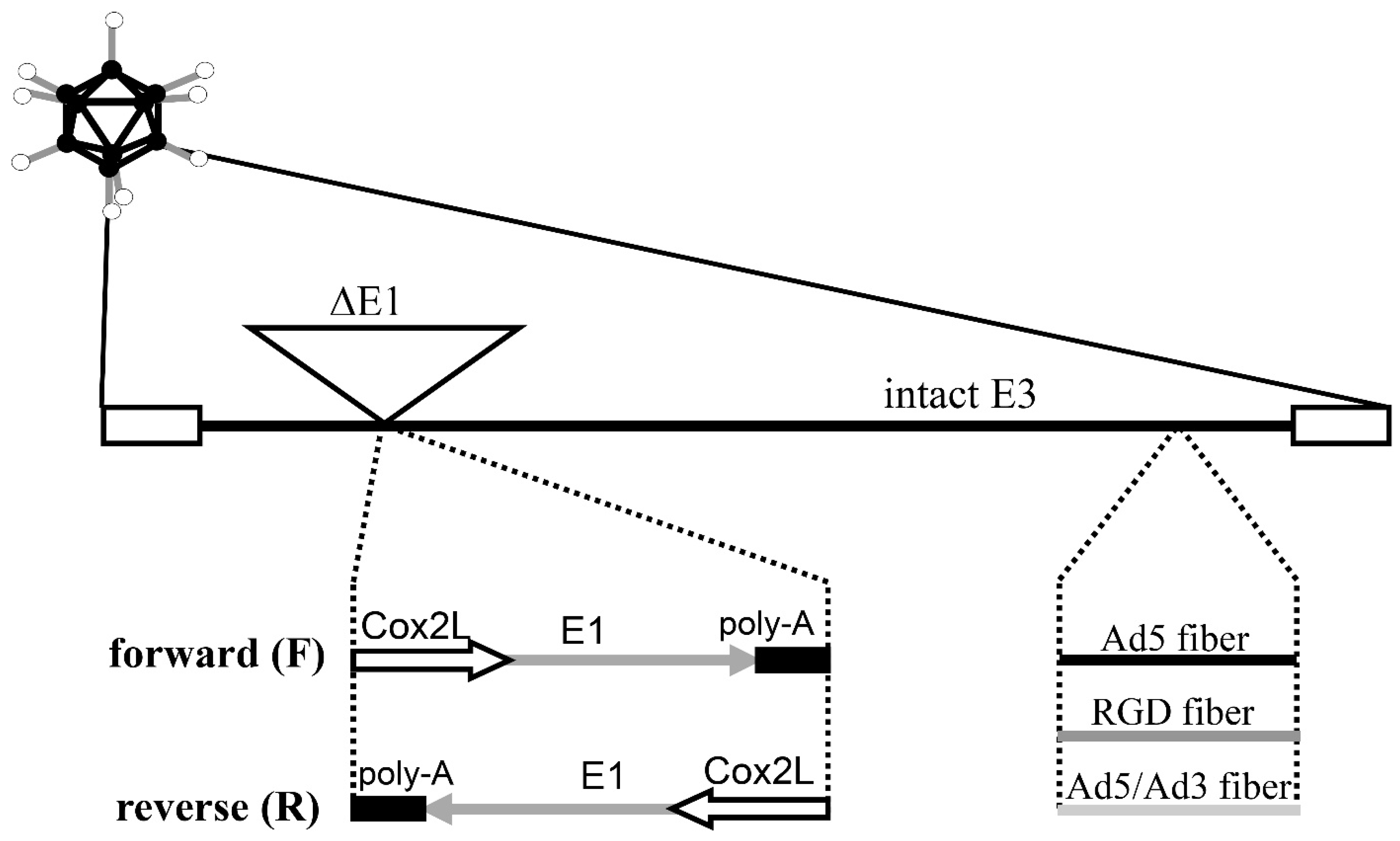
2.2. Adenoviral Vectors
2.3. Analysis of COX-2 RNA Levels
2.4. Promoter and Gene Delivery Analysis with Luciferase-Expressing Ads
2.5. In Vitro Analysis of Cytocidal Effect by Crystal Violet Staining
2.6. Quantitative In Vitro Cytotoxicity Assay
2.7. In Vivo Antitumor Effect in a Prostate Cancer Xenograft Model
2.8. Statistical Methods
3. Results
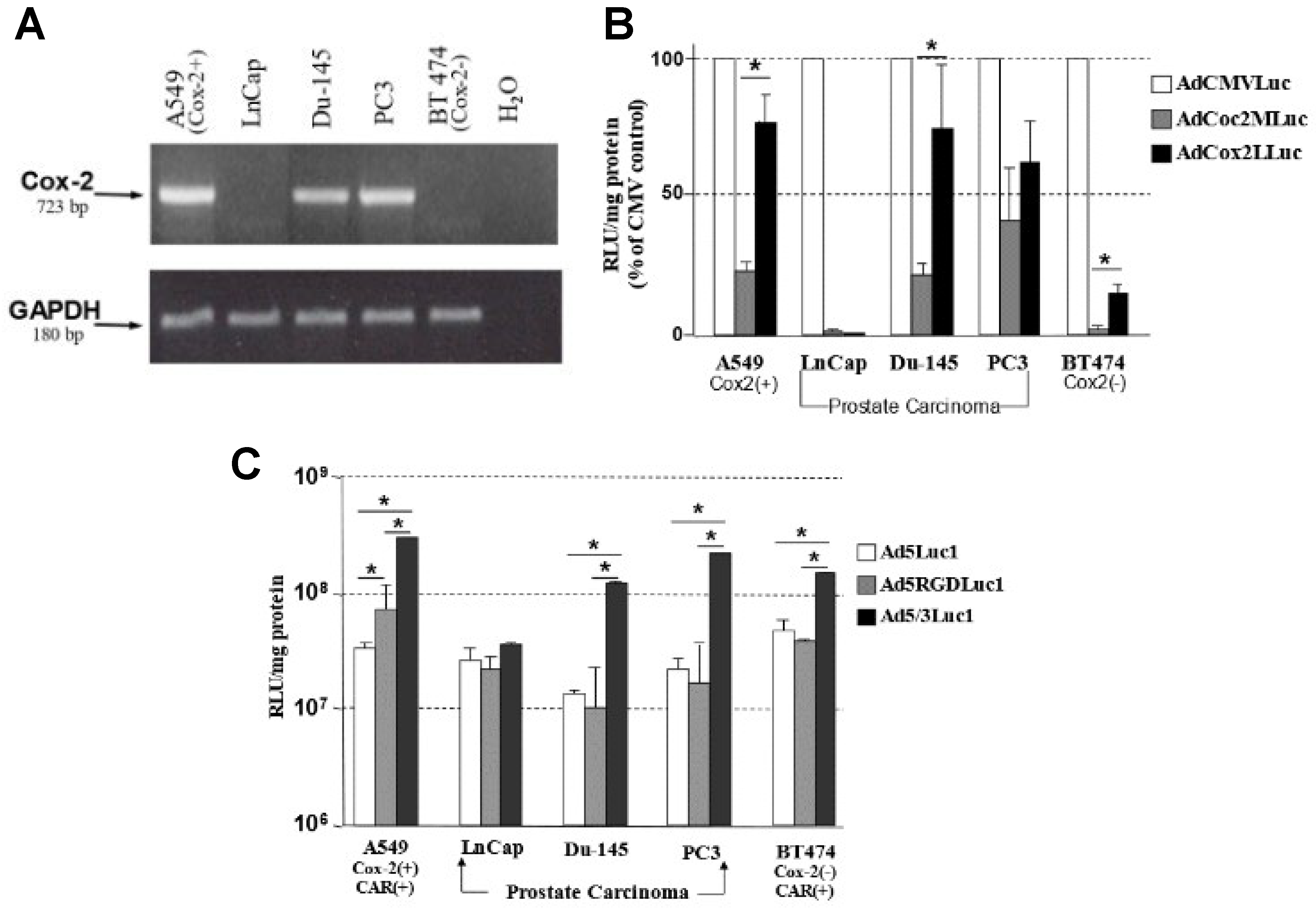
3.1. Transcriptional Status of COX-2 in Prostate Carcinoma Cell Lines
3.2. The Selectivity of the COX-2 Promoter for Prostate Cancer
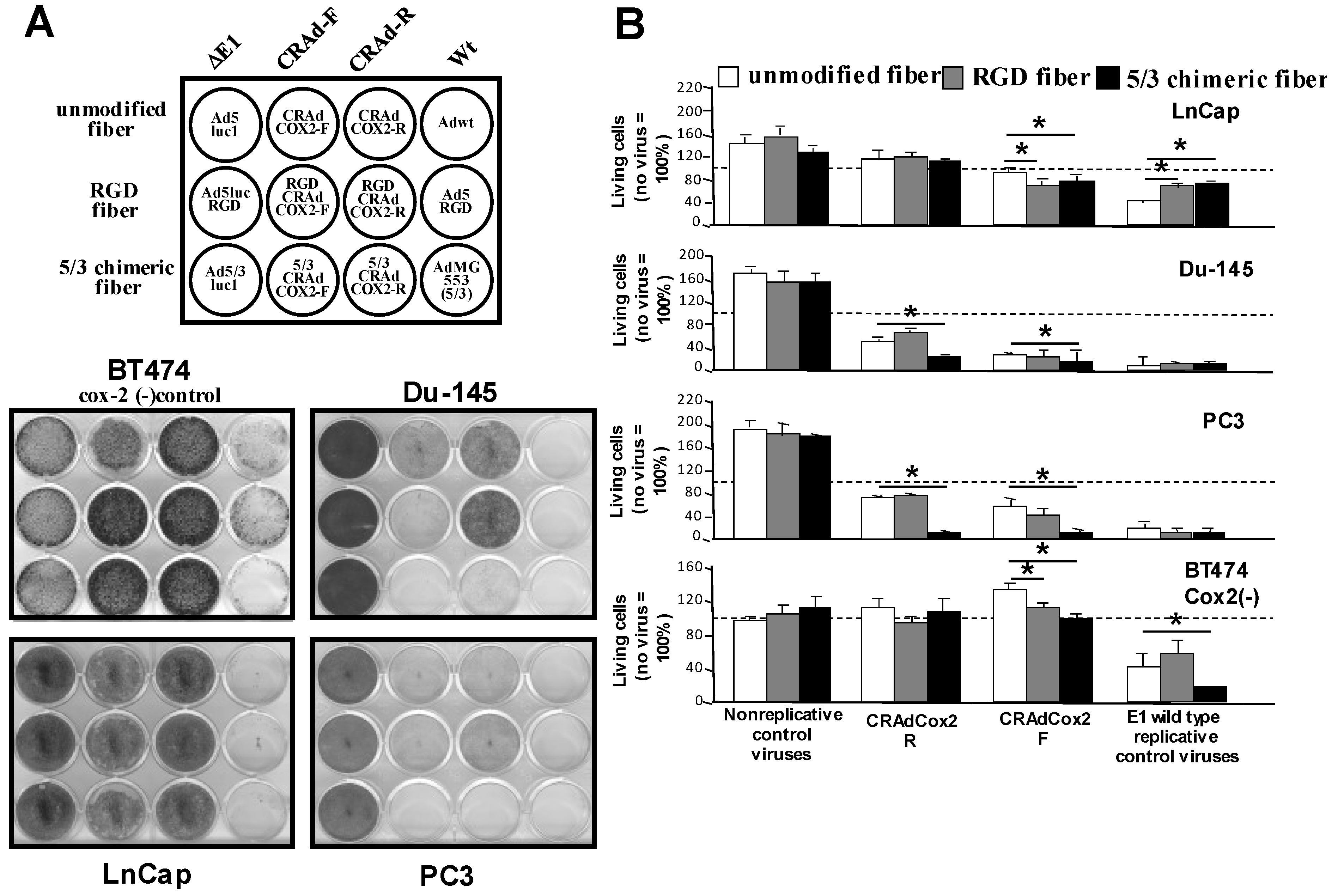
3.3. Analysis of Transduction Efficiency of RGD- and 5/3-Modified Vectors in Prostate Cancer
3.4. Increased Oncolytic Efficiency of Infectivity-Enhanced, COX-2 CRAds In Vitro
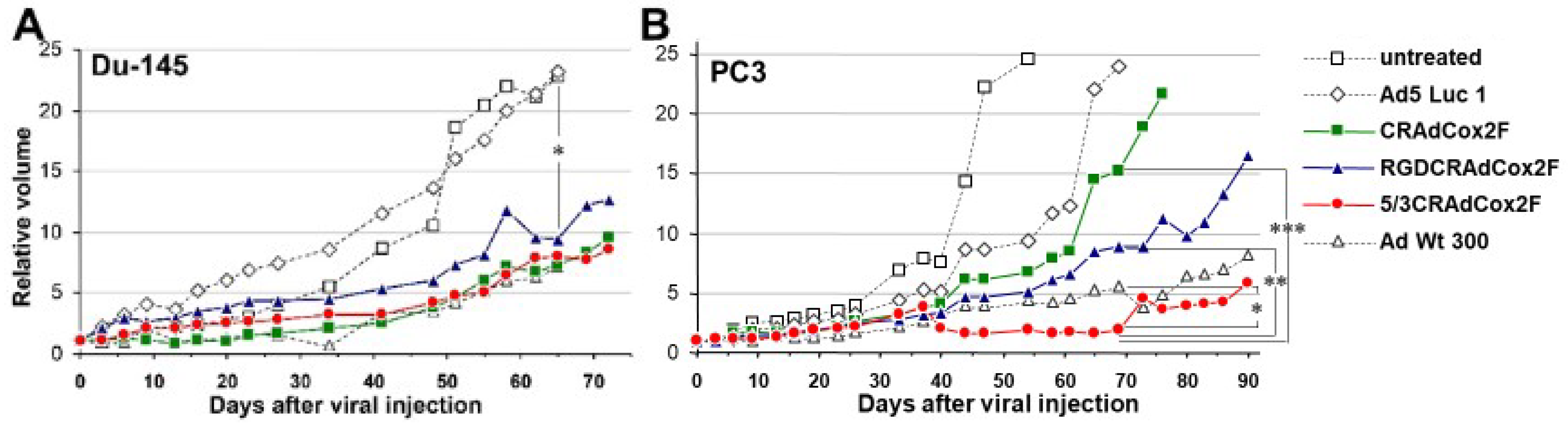
3.5. Therapeutic Efficacy of Infectivity-Enhanced CRAds In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Paquette, E.L.; Sun, L.; Paquette, L.R.; Connelly, R.; McLeod, D.G.; Moul, J.W. Improved prostate cancer-specific survival and other disease parameters: Impact of prostate-specific antigen testing. Urology 2002, 60, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Eisenberger, M.A. Expanding treatment options for metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 2055–2058. [Google Scholar] [CrossRef] [Green Version]
- Hirst, C.J.; Cabrera, C.; Kirby, M. Epidemiology of castration resistant prostate cancer: A longitudinal analysis using a UK primary care database. Cancer Epidemiol. 2012, 36, e349–e353. [Google Scholar] [CrossRef]
- Mansinho, A.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. Adv. Exp. Med. Biol. 2018, 1096, 117–133. [Google Scholar] [CrossRef] [PubMed]
- EAU Guidelines: Prostate Cancer. EAU Guidelines. Edn. Presented at the EAU Annual Congress Amsterdam 2022. 2022. Available online: https://uroweb.org/guideline/prostate-cancer/#6_4 (accessed on 25 January 2023).
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Hsu, E.C.; Shen, M.; Aslan, M.; Liu, S.; Kumar, M.; Garcia-Marques, F.; Nguyen, H.M.; Nolley, R.; Pitteri, S.J.; Corey, E.; et al. MCM2-7 complex is a novel druggable target for neuroendocrine prostate cancer. Sci. Rep. 2021, 11, 13305. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Cyclooxygenase-2: A potential target in breast cancer. Semin. Oncol. 2004, 31 (Suppl. 3), 64–73. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; DuBois, R.N. Cyclooxygenase-2 and gastrointestinal cancer. Cancer J. 2004, 10, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, A.; Liu, X.; Yao, S.; Levine, A.C. The role of cyclooxygenase-2 in prostate cancer. Urology 2001, 58 (Suppl. 1), 127–131. [Google Scholar] [CrossRef] [PubMed]
- DuBois, R.N. Cyclooxygenase-2 and colorectal cancer. Prog. Exp. Tumor Res. 2003, 37, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; DuBois, R.N. Cyclooxygenase as a target in lung cancer. Clin. Cancer Res. 2004, 10, 4266s–4269s. [Google Scholar] [CrossRef]
- Khor, L.Y.; Bae, K.; Pollack, A.; Hammond, M.E.; Grignon, D.J.; Venkatesan, V.M.; Rosenthal, S.A.; Ritter, M.A.; Sandler, H.M.; Hanks, G.E.; et al. COX-2 expression predicts prostate-cancer outcome: Analysis of data from the RTOG 92-02 trial. Lancet Oncol. 2007, 8, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Bonkhoff, H.; Berges, R. From pathogenesis to prevention of castration resistant prostate cancer. Prostate 2010, 70, 100–112. [Google Scholar] [CrossRef]
- Mitsunari, K.; Miyata, Y.; Asai, A.; Matsuo, T.; Shida, Y.; Hakariya, T.; Sakai, H. Human antigen R is positively associated with malignant aggressiveness via upregulation of cell proliferation, migration, and vascular endothelial growth factors and cyclooxygenase-2 in prostate cancer. Transl. Res. 2016, 175, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, B.A.; Reddy, B.S.; Bosland, M.C.; Nargi, D.; Horton, L.; Randolph, C.; Narayanan, N.K. Exisulind in combination with celecoxib modulates epidermal growth factor receptor, cyclooxygenase-2, and cyclin D1 against prostate carcinogenesis: In vivo evidence. Clin. Cancer Res. 2007, 13, 5965–5973. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Lee, R.; Holland, J.F.; Levine, A.C. Inhibition of cyclooxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. J. Urol. 2000, 164, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Mantwill, K.; Klein, F.G.; Wang, D.; Hindupur, S.V.; Ehrenfeld, M.; Holm, P.S.; Nawroth, R. Concepts in Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2021, 22, 10522. [Google Scholar] [CrossRef]
- Yamamoto, M.; Curiel, D.T. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Davydova, J.; Wang, M.; Siegal, G.P.; Krasnykh, V.; Vickers, S.M.; Curiel, D.T. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology 2003, 125, 1203–1218. [Google Scholar] [CrossRef]
- Yamamoto, M.; Alemany, R.; Adachi, Y.; Grizzle, W.E.; Curiel, D.T. Characterization of the cyclooxygenase-2 promoter in an adenoviral vector and its application for the mitigation of toxicity in suicide gene therapy of gastrointestinal cancers. Mol. Ther. 2001, 3, 385–394. [Google Scholar] [CrossRef]
- Davydova, J.; Le, L.P.; Gavrikova, T.; Wang, M.; Krasnykh, V.; Yamamoto, M. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004, 64, 4319–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Shenk, T. Adenoviridae: The Visuses and Their Replication. In Fields Virology; Fields, B., Knipe, D., Howley, P., Eds.; Lipponcott-Raven: Philadelphia, PA, USA, 1996; Volume 2, pp. 2111–2148. [Google Scholar]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [CrossRef] [Green Version]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Inoue, H.; Kosaka, T.; Miyata, A.; Hara, S.; Yokoyama, C.; Nanayama, T.; Tanabe, T. Structure and expression of the human prostaglandin endoperoxide synthase 2 gene. Adv. Prostaglandin. Thromboxane Leukot. Res. 1995, 23, 109–111. [Google Scholar]
- Inoue, H.; Nanayama, T.; Hara, S.; Yokoyama, C.; Tanabe, T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994, 350, 51–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Yokoyama, C.; Hara, S.; Tone, Y.; Tanabe, T. Transcriptional regulation of human prostaglandin-endoperoxide synthase- 2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J. Biol. Chem. 1995, 270, 24965–24971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnykh, V.; Dmitriev, I.; Mikheeva, G.; Miller, C.R.; Belousova, N.; Curiel, D.T. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J. Virol. 1998, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Krasnykh, V.N.; Mikheeva, G.V.; Douglas, J.T.; Curiel, D.T. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J. Virol. 1996, 70, 6839–6846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Curiel, D.T. Nonreplicating DNA viral vectors for suicide gene therapy: The adenoviral vectors. Methods Mol. Med. 2004, 90, 61–70. [Google Scholar]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Lorente, D.; Mateo, J.; Perez-Lopez, R.; de Bono, J.S.; Attard, G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol. 2015, 16, e279–e292. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar]
- Lee, S.J.; Kim, H.S.; Yu, R.; Lee, K.; Gardner, T.A.; Jung, C.; Jeng, M.H.; Yeung, F.; Cheng, L.; Kao, C. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol. Ther. 2002, 6, 415–421. [Google Scholar] [CrossRef]
- Dube, J.Y.; Chapdelaine, P.; Guerin, S.; Leclerc, S.; Rennie, P.S.; Matusik, R.J.; Tremblay, R.R. Search for androgen response elements in the proximal promoter of the canine prostate arginine esterase gene. J. Androl. 1995, 16, 304–311. [Google Scholar] [PubMed]
- O’Keefe, D.S.; Su, S.L.; Bacich, D.J.; Horiguchi, Y.; Luo, Y.; Powell, C.T.; Zandvliet, D.; Russell, P.J.; Molloy, P.L.; Nowak, N.J.; et al. Mapping, genomic organization and promoter analysis of the human prostate-specific membrane antigen gene. Biochim. Biophys. Acta 1998, 1443, 113–127. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.B.; Pearson, J.D.; Metter, E.J.; Brant, L.J.; Chan, D.W.; Andres, R.; Fozard, J.L.; Walsh, P.C. Longitudinal evaluation of prostate-specific antigen levels in men with and without prostate disease. JAMA 1992, 267, 2215–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef]
- Tilley, W.D.; Buchanan, G.; Hickey, T.E.; Bentel, J.M. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin. Cancer Res. 1996, 2, 277–285. [Google Scholar]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Wei, F.K.; Xu, Z.Y.; Wen, R.M.; Chen, J.C.; Wang, J.Q.; Mao, L.J. Tropism and transduction of oncolytic adenovirus vectors in prostate cancer therapy. Front. Biosci. (Landmark Ed.) 2021, 26, 866–872. [Google Scholar] [CrossRef]
- Yoshimura, R.; Sano, H.; Masuda, C.; Kawamura, M.; Tsubouchi, Y.; Chargui, J.; Yoshimura, N.; Hla, T.; Wada, S. Expression of cyclooxygenase-2 in prostate carcinoma. Cancer 2000, 89, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, H.Y.; Yuan, T.; Luo, J.; Zhou, W.D.; Jiang, Q.Q.; Wu, D. Enzalutamide-Induced Upregulation of PCAT6 Promotes Prostate Cancer Neuroendocrine Differentiation by Regulating miR-326/HNRNPA2B1 Axis. Front. Oncol. 2021, 11, 650054. [Google Scholar] [CrossRef] [PubMed]
- Spetsieris, N.; Boukovala, M.; Patsakis, G.; Alafis, I.; Efstathiou, E. Neuroendocrine and Aggressive-Variant Prostate Cancer. Cancers 2020, 12, 3792. [Google Scholar] [CrossRef]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Umesono, K.; Nishimori, T.; Hirata, Y.; Tanabe, T. Glucocorticoid-mediated suppression of the promoter activity of the cyclooxygenase-2 gene is modulated by expression of its receptor in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 254, 292–298. [Google Scholar] [CrossRef]
- Raj, V.; Bhadauria, A.S.; Singh, A.K.; Kumar, U.; Rai, A.; Keshari, A.K.; Kumar, P.; Kumar, D.; Maity, B.; Nath, S.; et al. Novel 1,3,4-thiadiazoles inhibit colorectal cancer via blockade of IL-6/COX-2 mediated JAK2/STAT3 signals as evidenced through data-based mathematical modeling. Cytokine 2019, 118, 144–159. [Google Scholar] [CrossRef]
- Watanabe, Y.; Imanishi, Y.; Ozawa, H.; Sakamoto, K.; Fujii, R.; Shigetomi, S.; Habu, N.; Otsuka, K.; Sato, Y.; Sekimizu, M.; et al. Selective EP2 and Cox-2 inhibition suppresses cell migration by reversing epithelial-to-mesenchymal transition and Cox-2 overexpression and E-cadherin downregulation are implicated in neck metastasis of hypopharyngeal cancer. Am. J. Transl. Res. 2020, 12, 1096–1113. [Google Scholar]
- Majumder, M.; Landman, E.; Liu, L.; Hess, D.; Lala, P.K. COX-2 Elevates Oncogenic miR-526b in Breast Cancer by EP4 Activation. Mol. Cancer Res. 2015, 13, 1022–1033. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrikova, T.; Nakamura, N.; Davydova, J.; Antonarakis, E.S.; Yamamoto, M. Infectivity-Enhanced, Conditionally Replicative Adenovirus for COX-2-Expressing Castration-Resistant Prostate Cancer. Viruses 2023, 15, 901. https://doi.org/10.3390/v15040901
Gavrikova T, Nakamura N, Davydova J, Antonarakis ES, Yamamoto M. Infectivity-Enhanced, Conditionally Replicative Adenovirus for COX-2-Expressing Castration-Resistant Prostate Cancer. Viruses. 2023; 15(4):901. https://doi.org/10.3390/v15040901
Chicago/Turabian StyleGavrikova, Tatyana, Naohiko Nakamura, Julia Davydova, Emmanuel S. Antonarakis, and Masato Yamamoto. 2023. "Infectivity-Enhanced, Conditionally Replicative Adenovirus for COX-2-Expressing Castration-Resistant Prostate Cancer" Viruses 15, no. 4: 901. https://doi.org/10.3390/v15040901
APA StyleGavrikova, T., Nakamura, N., Davydova, J., Antonarakis, E. S., & Yamamoto, M. (2023). Infectivity-Enhanced, Conditionally Replicative Adenovirus for COX-2-Expressing Castration-Resistant Prostate Cancer. Viruses, 15(4), 901. https://doi.org/10.3390/v15040901