
CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120

Abstract
:
{kind=link}
{kind=link}
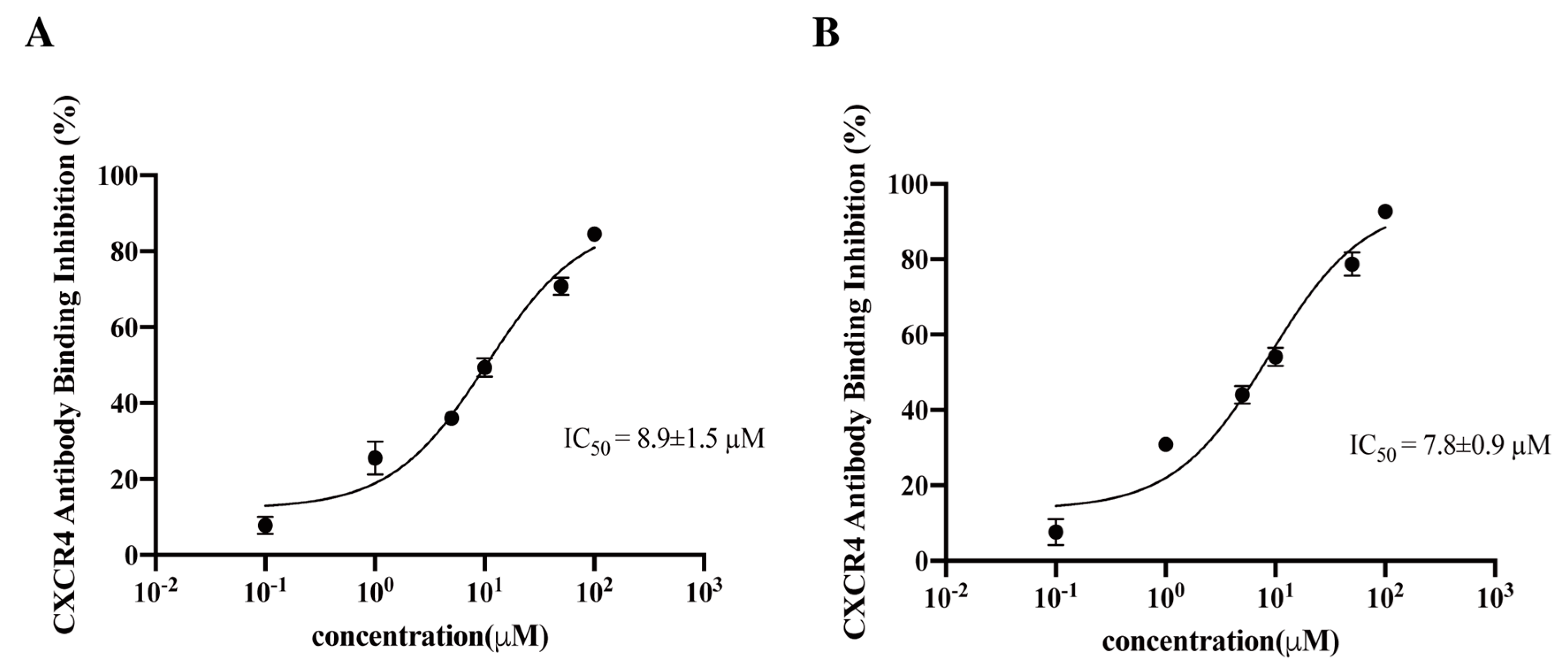{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
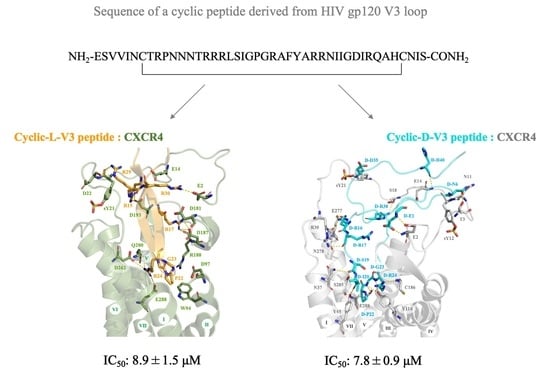
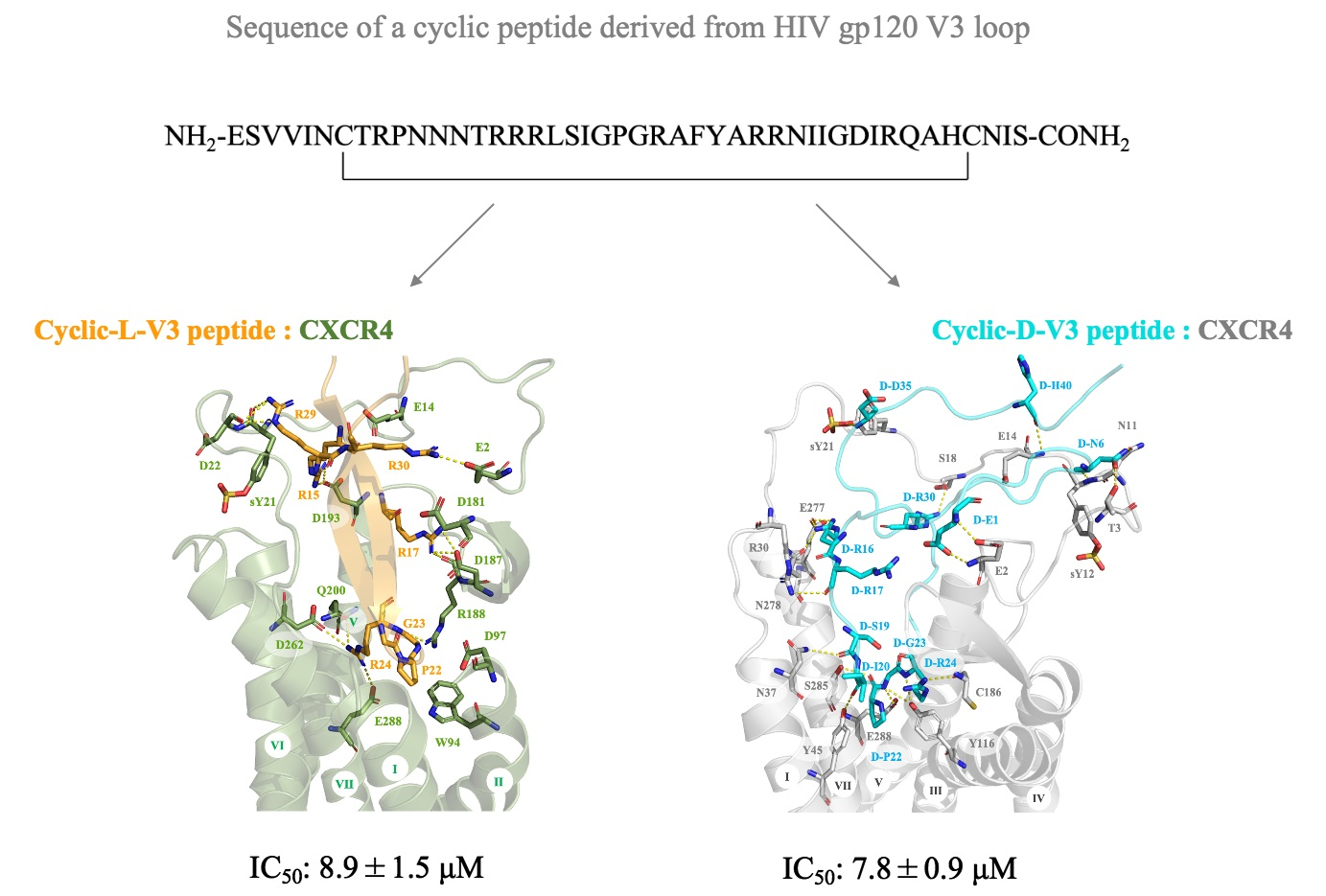
2.1. Design of Cyclic L- and D-Peptides Corresponding to the Full-Length V3 Loop of gp120 of the Dual-Tropic HIV 89.6 Strain
2.2. Molecular Modeling of CXCR4 Interactions with Cyclic L- and D-V3 Loop Peptides
3. Conclusions
3.1. Experimental Procedures
3.1.1. Peptide Synthesis
3.1.2. Competitive Binding Assay
3.1.3. Peptide-CXCR4 Docking
3.1.4. MD Simulation
3.1.5. Binding Free Energy Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, W.T.; Yang, Y.L.; Xu, Y.; An, J. Targeting Chemokine Receptor CXCR4 for Treatment of HIV-1 Infection, Tumor Progression, and Metastasis. Curr. Top. Med. Chem. 2014, 14, 1574–1589. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Farzan, M.; Choe, H.; Parolin, C.; ClarkLewis, I.; Sodroski, J.; Springer, T.A. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996, 382, 829–833. [Google Scholar] [CrossRef]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996, 382, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Mazo, I.B.; Massberg, S.; von Andrian, U.H. Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 2011, 32, 493–503. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Saini, V.; Marchese, A.; Majetschak, M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 2010, 285, 15566–15576. [Google Scholar] [CrossRef]
- Saini, V.; Marchese, A.; Tang, W.J.; Majetschak, M. Structural determinants of ubiquitin-CXC chemokine receptor 4 interaction. J. Biol. Chem. 2011, 286, 44145–44152. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef]
- Trkola, A.; Dragic, T.; Arthos, J.; Binley, J.M.; Olson, W.C.; Allaway, G.P.; Cheng-Mayer, C.; Robinson, J.; Maddon, P.J.; Moore, J.P. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 1996, 384, 184–187. [Google Scholar] [CrossRef]
- Maddon, P.J.; Dalgleish, A.G.; McDougal, J.S.; Clapham, P.R.; Weiss, R.A.; Axel, R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986, 47, 333–348. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Doms, R.W.; Fenyo, E.M.; Korber, B.T.; Littman, D.R.; Moore, J.P.; Sattentau, Q.J.; Schuitemaker, H.; Sodroski, J.; Weiss, R.A. A new classification for HIV-1. Nature 1998, 391, 240. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, A.; Hosoya, N.; Kawana-Tachikawa, A. HIV-1 tropism. Protein Cell 2010, 1, 510–513. [Google Scholar] [CrossRef]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef]
- Choi, W.T.; Duggineni, S.; Xu, Y.; Huang, Z.; An, J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4). J. Med. Chem. 2012, 55, 977–994. [Google Scholar] [CrossRef]
- Zhou, N.M.; Luo, Z.W.; Luo, J.S.; Fan, X.J.; Cayabyab, M.; Hiraoka, M.; Liu, D.X.; Han, X.B.; Pesavento, J.; Dong, C.Z.; et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J. Biol. Chem. 2002, 277, 17476–17485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, L.S.; Zhu, R.; Meng, Q.; Zhu, S.; Xu, Y.; Zhang, H.; Fang, X.; Zhang, X.; Zhou, J.; et al. High affinity CXCR4 inhibitors generated by linking low affinity peptides. Eur. J. Med. Chem. 2019, 172, 174–185. [Google Scholar] [CrossRef]
- Zhu, R.; Meng, Q.; Zhang, H.; Zhang, G.; Huang, L.S.M.; Xu, Y.; Schooley, R.T.; An, J.; Huang, Z. HIV-1 gp120-CXCR4 recognition probed with synthetic nanomolar affinity D-peptides containing fragments of gp120 V3 loop. Eur. J. Med. Chem. 2022, 224, 114797. [Google Scholar] [CrossRef]
- Xu, Y.; Duggineni, S.; Espitia, S.; Richman, D.D.; An, J.; Huang, Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem. Biophys. Res. Commun. 2013, 435, 646–650. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, M.; Zhang, Q.; Zhang, C.; Yang, X.; Huang, Z.; An, J. Design, synthesis, and biological characterization of novel PEG-linked dimeric modulators for CXCR4. Bioorg. Med. Chem. 2016, 24, 5393–5399. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. 1), S162–S173. [Google Scholar] [CrossRef]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef]
- Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Conway, P.; Tyka, M.D.; DiMaio, F.; Konerding, D.E.; Baker, D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014, 23, 47–55. [Google Scholar] [CrossRef]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 2010, 78, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Ng, Y.K. Calibur: A tool for clustering large numbers of protein decoys. BMC Bioinform. 2010, 11, 25. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Singh, N.; Warshel, A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins 2010, 78, 1705–1723. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.; Sang, X.; Zhou, J.; Meng, Q.; Huang, L.S.M.; Xu, Y.; An, J.; Huang, Z. CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120. Viruses 2023, 15, 1084. https://doi.org/10.3390/v15051084
Zhu R, Sang X, Zhou J, Meng Q, Huang LSM, Xu Y, An J, Huang Z. CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120. Viruses. 2023; 15(5):1084. https://doi.org/10.3390/v15051084
Chicago/Turabian StyleZhu, Ruohan, Xiaohong Sang, Jiao Zhou, Qian Meng, Lina S. M. Huang, Yan Xu, Jing An, and Ziwei Huang. 2023. "CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120" Viruses 15, no. 5: 1084. https://doi.org/10.3390/v15051084
APA StyleZhu, R., Sang, X., Zhou, J., Meng, Q., Huang, L. S. M., Xu, Y., An, J., & Huang, Z. (2023). CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120. Viruses, 15(5), 1084. https://doi.org/10.3390/v15051084