
African Swine Fever Virus Interaction with Host Innate Immune Factors


Abstract
:1. Introduction
2. ASFV Proteins and Innate Immunity
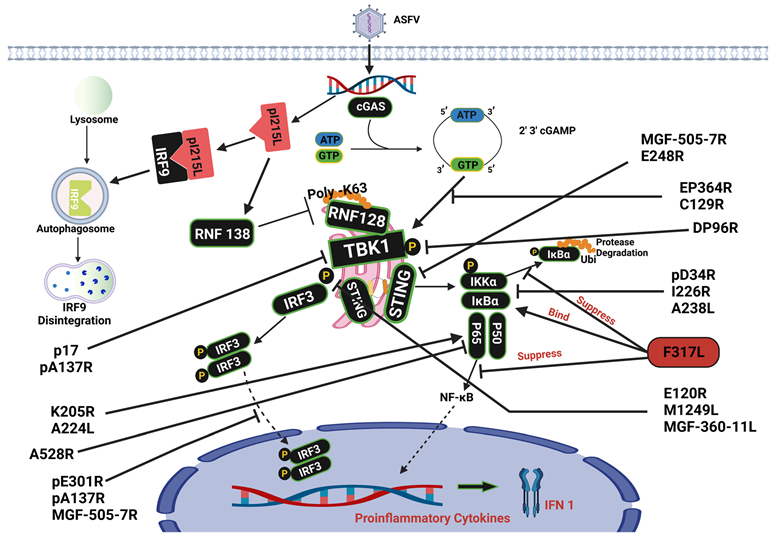
3. Cyclic GMP-AMP Synthase Signaling Pathway
4. NF-κB Signaling Pathway
5. Transforming Growth Factor-β Signaling Pathway
6. Ubiquitination
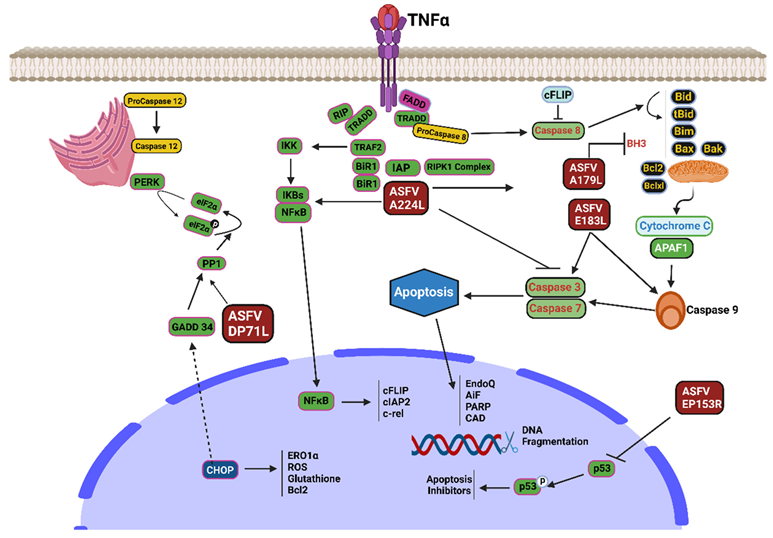
7. ASFV Modulating Apoptosis Protein
8. Inhibition of Apoptosis
8.1. A179L Protein
8.2. A224L Protein
8.3. EP153R
8.4. DP71L
9. Ornithodoros Tick Antiviral Response to ASFV
10. Warthog Resistance to ASFV
11. Discussion and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallardo, C.; Soler, A.; Nieto, R.; Sánchez, M.A.; Martins, C.; Pelayo, V.; Carrascosa, A.; Revilla, Y.; Simón, A.; Briones, V.; et al. Experimental Transmission of African Swine Fever (ASF) Low Virulent Isolate NH/P68 by Surviving Pigs. Transbound. Emerg. Dis. 2015, 62, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular characterization of African swine fever virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, S.; Williamson, A.; Malesa, R.; Van Heerden, J.; Boshoff, C.I.; Bastos, A.D.S.; Heath, L.; Carulei, O. Genome Sequences of Three African Swine Fever Viruses of Genotypes I, III, and XXII from South Africa and Zambia, Isolated from Ornithodoros Soft Ticks. Microbiol. Resour. Announc. 2020, 9, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Urbano, A.C.; Ferreira, F. Role of the dna-binding protein pa104r in asfv genome packaging and as a novel target for vaccine and drug development. Vaccines 2020, 8, 585. [Google Scholar] [CrossRef]
- Franzoni, G.; Zinellu, S.; Carta, T.; De Ciucis, C.G.; Fruscione, F.; Anfossi, A.; Ledda, M.; Graham, S.P.; Dei Giudici, S.; Razzuoli, E.; et al. Analyses of the Impact of Immunosuppressive Cytokines on Porcine Macrophage Responses and Susceptibility to Infection to African Swine Fever Viruses. Pathogens 2022, 11, 166. [Google Scholar] [CrossRef]
- Dixon, L.K.; Islam, M.; Nash, R.; Reis, A.L. African swine fever virus evasion of host defences. Virus Res. 2019, 266, 25–33. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Ye, G.; Xue, M.; Yu, H.; Feng, C.; Zhou, Q.; Liu, X.; Zhang, L.; Jiao, S.; et al. African swine fever virus pE301R negatively regulates cGAS-STING signaling pathway by inhibiting the nuclear translocation of IRF3. Vet. Microbiol. 2022, 274, 109556. [Google Scholar] [CrossRef]
- Franzoni, G.; Graham, S.P.; Giudici, S.D.; Bonelli, P.; Pilo, G.; Anfossi, A.G.; Pittau, M.; Nicolussi, P.S.; Laddomada, A.; Oggiano, A. Characterization of the interaction of African swine fever virus with monocytes and derived macrophage subsets. Vet. Microbiol. 2017, 198, 88–98. [Google Scholar] [CrossRef]
- Satoh, T.; Akira, S. Toll-like receptor signaling and its inducible proteins. In Myeloid Cells in Health and Disease: A Synthesis; Wiley: Hoboken, NJ, USA, 2017; pp. 447–453. [Google Scholar] [CrossRef]
- Montoya, M.; Franzoni, G.; Revilla, Y.; Galindo, I.; Alonso, C. Understanding and Combatting African Swine Fever; Wageningen Academic: Wageningen, The Netherland, 2021; pp. 63–85. [Google Scholar] [CrossRef]
- Li, J.; Song, J.; Kang, L.; Huang, L.; Zhou, S.; Hu, L.; Zheng, J.; Li, C.; Zhang, X.; He, X.; et al. pMGF505-7R determines pathogenicity of African swine fever virus infection by inhibiting IL-1β and type I IFN production. PLoS Pathog. 2021, 17, e1009733. [Google Scholar] [CrossRef]
- Sánchez, E.G.; Quintas, A.; Nogal, M.; Castelló, A.; Revilla, Y. African swine fever virus controls the host transcription and cellular machinery of protein synthesis. Virus Res. 2013, 173, 58–75. [Google Scholar] [CrossRef]
- Dodantenna, N.; Ranathunga, L.; Chathuranga, W.A.G.; Weerawardhana, A.; Cha, J.-W.; Subasinghe, A.; Gamage, N.; Haluwana, D.K.; Kim, Y.; Jheong, W.; et al. African Swine Fever Virus EP364R and C129R Target Cyclic GMP-AMP To Inhibit the cGAS-STING Signaling Pathway. J. Virol. 2022, 96, e01022-22. [Google Scholar] [CrossRef]
- Sun, M.; Yu, S.; Ge, H.; Wang, T.; Li, Y.; Zhou, P.; Pan, L.; Han, Y.; Yang, Y.; Sun, Y.; et al. The A137R Protein of African Swine Fever Virus Inhibits Type I Interferon Production via the Autophagy-Mediated Lysosomal Degradation of TBK1. J. Virol. 2022, 96, e01957-21. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Gao, X.; Xin, T.; Wang, X.; Yu, H.; Chen, S.; Jiang, Y.; Chen, Q.; Jiang, F.; et al. African swine fever virus M1249L protein antagonizes type I interferon production via suppressing phosphorylation of TBK1 and degrading IRF3. Virus Res. 2022, 319, 198872. [Google Scholar] [CrossRef]
- Zheng, W.; Xia, N.; Zhang, J.; Cao, Q.; Jiang, S.; Luo, J.; Wang, H.; Chen, N.; Zhang, Q.; Meurens, F.; et al. African Swine Fever Virus Structural Protein p17 Inhibits cGAS-STING Signaling Pathway through Interacting with STING. Front. Immunol. 2022, 13, 941579. [Google Scholar] [CrossRef]
- Yang, K.; Xue, Y.; Niu, H.; Shi, C.; Cheng, M.; Wang, J.; Zou, B.; Wang, J.; Niu, T.; Bao, M.; et al. African swine fever virus MGF360-11L negatively regulates cGAS-STING-mediated inhibition of type I interferon production. Vet. Res. 2022, 53, 7. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Z.; Feng, T.; Ma, Z.; Xue, Q.; Wu, P.; Li, P.; Li, S.; Yang, F.; Cao, W.; et al. African Swine Fever Virus E120R Protein Inhibits Interferon Beta Production by Interacting with IRF3 To Block Its Activation. J. Virol. 2021, 95, e00824-21. [Google Scholar] [CrossRef]
- Yang, K.; Huang, Q.; Wang, R.; Zeng, Y.; Cheng, M.; Xue, Y.; Shi, C.; Ye, L.; Yang, W.; Jiang, Y.; et al. African swine fever virus MGF505-11R inhibits type I interferon production by negatively regulating the cGAS-STING-mediated signaling pathway. Vet. Microbiol. 2021, 263, 109265. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Z.; Gao, X.; Lv, J.; Hu, Y.; Jung, Y.S.; Zhu, S.; Wu, X.; Qian, Y.; Dai, J. ASFV pD345L protein negatively regulates NF-κB signalling by inhibiting IKK kinase activity. Vet. Res. 2022, 53, 32. [Google Scholar] [CrossRef]
- Hong, J.; Chi, X.; Yuan, X.; Wen, F.; Rai, K.R.; Wu, L.; Song, Z.; Wang, S.; Guo, G.; Chen, J.L. I226R Protein of African Swine Fever Virus Is a Suppressor of Innate Antiviral Responses. Viruses 2022, 14, 575. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; Feng, T.; Zhang, X.; Yang, F.; Cao, W.; Chen, H.; Liu, H.; Zhang, K.; Zhu, Z.; et al. African Swine Fever Virus F317L Protein Inhibits NF-κB Activation To Evade Host Immune Response and Promote Viral Replication. mSphere 2021, 6, e00658-21. [Google Scholar] [CrossRef]
- Liu, X.; Ao, D.; Jiang, S.; Xia, N.; Xu, Y.; Shao, Q.; Luo, J.; Wang, H.; Zheng, W.; Chen, N.; et al. African swine fever virus a528r inhibits tlr8 mediated nf-κb activity by targeting p65 activation and nuclear translocation. Viruses 2021, 13, 2046. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, W.; Liu, H.; Xue, M.; Liu, X.; Zhang, K.; Hu, L.; Li, J.; Liu, X.; Xiang, Z.; et al. African Swine Fever Virus pI215L Negatively Regulates cGAS-STING Signaling Pathway through Recruiting RNF138 to Inhibit K63-Linked Ubiquitination of TBK1. J. Immunol. 2021, 207, 2754–2769. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Nie, Y.; Li, H.; Chen, W.; Lin, W.; Chen, F.; Xie, Q. African Swine Fever Virus Protein E199L Promotes Cell Autophagy through the Interaction of PYCR2. Virol. Sin. 2021, 36, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, L.; Wang, J.; Su, D.; Li, D.; Du, Y.; Yang, G.; Zhang, G.; Chu, B. African Swine Fever Virus K205R Induces ER Stress and Consequently Activates Autophagy and the NF-κB Signaling Pathway. Viruses 2022, 14, 394. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Cabezas, M.; Munoz-Moreno, R.; Galindo, I.; Cuesta-Geijo, M.A.; Alonso, C. A179L, a New Viral Bcl2 Homolog Targeting Beclin 1 Autophagy Related Protein. Curr. Mol. Med. 2013, 13, 305–316. [Google Scholar] [CrossRef]
- Banjara, S.; Caria, S.; Dixon, L.K.; Hinds, M.G.; Kvansakul, M. Structural Insight into African Swine Fever Virus A179L-Mediated Inhibition of Apoptosis. J. Virol. 2017, 91, e02228-16. [Google Scholar] [CrossRef]
- Shi, J.; Liu, W.; Zhang, M.; Sun, J.; Xu, X. The a179l gene of african swine fever virus suppresses virus-induced apoptosis but enhances necroptosis. Viruses 2021, 13, 2490. [Google Scholar] [CrossRef]
- Nogal, M.L.; González de Buitrago, G.; Rodríguez, C.; Cubelos, B.; Carrascosa, A.L.; Salas, M.L.; Revilla, Y. African Swine Fever Virus IAP Homologue Inhibits Caspase Activation and Promotes Cell Survival in Mammalian Cells. J. Virol. 2001, 75, 2535–2543. [Google Scholar] [CrossRef]
- Hurtado, C.; Granja, A.G.; Bustos, M.J.; Nogal, M.L.; González De Buitrago, G.; De Yébenes, V.G.; Salas, M.L.; Revilla, Y.; Carrascosa, A.L. The C-type lectin homologue gene (EP153R) of African swine fever virus inhibits apoptosis both in virus infection and in heterologous expression. Virology 2004, 326, 160–170. [Google Scholar] [CrossRef]
- Petrovan, V.; Rathakrishnan, A.; Islam, M.; Goatley, L.C.; Moffat, K.; Sanchez-Cordon, P.J.; Reis, A.L.; Dixon, L.K. Role of African Swine Fever Virus Proteins EP153R and EP402R in Reducing Viral Persistence in Blood and Virulence in Pigs Infected with BeninΔDP148R. J. Virol. 2022, 96, e01340-21. [Google Scholar] [CrossRef]
- Barber, C.; Netherton, C.; Goatley, L.; Moon, A.; Goodbourn, S.; Dixon, L. Identification of residues within the African swine fever virus DP71L protein required for dephosphorylation of translation initiation factor eIF2α and inhibiting activation of pro-apoptotic CHOP. Virology 2017, 504, 107–113. [Google Scholar] [CrossRef]
- Silk, R.N.; Bowick, G.C.; Abrams, C.C.; Dixon, L.K. African swine fever virus A238L inhibitor of NF-κB and of calcineurin phosphatase is imported actively into the nucleus and exported by a CRM1-mediated pathway. J. Gen. Virol. 2007, 88, 411–419. [Google Scholar] [CrossRef]
- Hernáez, B.; Díaz-Gil, G.; García-Gallo, M.; Quetglas, J.I.; Rodríguez-Crespo, I.; Dixon, L.; Dixon, L.; Escribano, J.M.; Alonso, C. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004, 569, 224–228. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, W.; Wen, Y.; Niu, Q.; Yang, J.; Guan, G.; Yin, H.; Zheng, H.; Li, D.; Liu, Z. The E248R protein of African swine fever virus inhibits the cGAS-STING-mediated innate immunity. Shengwu Gongcheng Xuebao/Chin. J. Biotechnol. 2022, 38, 1837–1846. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, J.; Ni, J.; Jiang, S.; Xia, N.; Guo, Y.; Shao, Q.; Cao, Q.; Zheng, W.; Chen, N.; et al. The African swine fever virus protease pS273R inhibits DNA sensing cGAS-STING pathway by targeting IKKε. Virulence 2022, 13, 740–756. [Google Scholar] [CrossRef]
- Luo, W.W.; Li, S.; Li, C.; Lian, H.; Yang, Q.; Zhong, B.; Shu, H.B. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 2016, 17, 1057–1066. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef]
- Ayanwale, A.; Trapp, S.; Guabiraba, R.; Caballero, I.; Roesch, F. New Insights in the Interplay Between African Swine Fever Virus and Innate Immunity and Its Impact on Viral Pathogenicity. Front. Microbiol. 2022, 13, 958307. [Google Scholar] [CrossRef]
- Abe, T.; Marutani, Y.; Shoji, I. Cytosolic DNA-sensing immune response and viral infection. Microbiol. Immunol. 2019, 63, 51–64. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Wu, Y.; Chen, H.; Zhang, S.; Li, J.; Xin, T.; Jia, H.; Hou, S.; Jiang, Y.; et al. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 2018, 506, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shi, X.-J.; Sun, H.-W.; Chen, H.-J. Insights into African swine fever virus immunoevasion strategies. J. Integr. Agric. 2020, 19, 11–22. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Song, Y.; Li, Z.; Xue, Z.; Cao, W.; Liu, X.; Zheng, H. African Swine Fever Virus Regulates Host Energy and Amino Acid Metabolism To Promote Viral Replication. J. Virol. 2022, 96, e01919-21. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB signaling in macrophages: Dynamics, crosstalk, and signal integration. Front. Immunol. 2019, 10, 00705. [Google Scholar] [CrossRef]
- Ashall, L.; Horton, C.A.; Nelson, D.E.; Paszek, P.; Harper, C.V.; Sillitoe, K.; Ryan, S.; Europe PMC Funders Group. Pulsatile stimulation determines timing and specificity of NF- kappa B-dependent transcription. Science 2009, 324, 242–246. [Google Scholar] [CrossRef]
- Zhao, G.; Li, T.; Liu, X.; Zhang, T.; Zhang, Z.; Kang, L.; Song, J.; Zhou, S.; Chen, X.; Wang, X.; et al. African swine fever virus cysteine protease pS273R inhibits pyroptosis by noncanonically cleaving gasdermin D. J. Biol. Chem. 2022, 298, 101480. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, Y.; Feng, Y.; Quan, W.; Luo, Y.; Wang, H.; Zheng, J.; Chen, X.; Huang, Z.; Chen, X.; et al. Effects of the NF-κB Signaling Pathway Inhibitor BAY11-7082 in the Replication of ASFV. Viruses 2022, 14, 297. [Google Scholar] [CrossRef]
- Barrado-Gil, L.; Del Puerto, A.; Galindo, I.; Cuesta-Geijo, M.Á.; García-Dorival, I.; de Motes, C.M.; Alonso, C. African swine fever virus ubiquitin-conjugating enzyme is an immunomodulator targeting NF-κB activation. Viruses 2021, 13, 1160. [Google Scholar] [CrossRef]
- Abkallo, H.M.; Hemmink, J.D.; Oduor, B.; Khazalwa, E.M.; Svitek, N.; Assad-Garcia, N.; Khayumbi, J.; Fuchs, W.; Vashee, S.; Steinaa, L. Co-Deletion of A238L and EP402R Genes from a Genotype IX African Swine Fever Virus Results in Partial Attenuation and Protection in Swine. Viruses 2022, 14, 2024. [Google Scholar] [CrossRef]
- Dixon, L.K.; Sánchez-Cordón, P.J.; Galindo, I.; Alonso, C. Investigations of pro- and anti-apoptotic factors affecting African swine fever virus replication and pathogenesis. Viruses 2017, 9, 241. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Mirzaei, H.; Faghihloo, E. Viruses as key modulators of the TGF-β pathway; a double-edged sword involved in cancer. Rev. Med. Virol. 2018, 28, e1967. [Google Scholar] [CrossRef]
- Morishima, N.; Mizoguchi, I.; Takeda, K.; Mizuguchi, J.; Yoshimoto, T. TGF-β is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23. Biochem. Biophys. Res. Commun. 2009, 386, 105–110. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef]
- Wells, R.G.V. TGF-β signaling pathways. Am. J. Physiol.-Gastrointest. Liver Physiol. 2000, 279, 845–850. [Google Scholar] [CrossRef]
- Yuniarti, W.M.; Primarizky, H. Mini Review: Liver Fibrosis Mechanism. KnE Life Sci. 2017, 3, 327. [Google Scholar] [CrossRef]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef]
- O’Donnell, V.; Risatti, G.R.; Holinka, L.G.; Krug, P.W.; Carlson, J.; Velazquez-Salinas, L.; Azzinaro, P.A.; Gladue, D.P.; Borca, M.V. Simultaneous Deletion of the 9GL and UK Genes from the African Swine Fever Virus Georgia 2007 Isolate Offers Increased Safety and Protection against Homologous Challenge. J. Virol. 2017, 91, e01760-16. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Kozak, E.; Lyjak, M.; Pejsak, Z.; Niemczuk, K. Transcriptional immunoresponse of tissue-specific macrophages in swine after infection with African swine fever virus. Bull. Vet. Inst. Pulawy 2015, 59, 441–445. [Google Scholar] [CrossRef]
- Post, J.; Weesendorp, E.; Montoya, M.; Loeffen, W.L. Influence of Age and Dose of African Swine Fever Virus Infections on Clinical Outcome and Blood Parameters in Pigs. Viral Immunol. 2017, 30, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, J.; Zhang, Y.; Yang, J.; Wang, L.; Qi, Y.; Han, X.; Zhou, X.; Miao, F.; Chen, T.; et al. Cytokine Storm in Domestic Pigs Induced by Infection of Virulent African Swine Fever Virus. Front. Vet. Sci. 2021, 7, 601641. [Google Scholar] [CrossRef] [PubMed]
- Carta, T.; Razzuoli, E.; Fruscione, F.; Zinellu, S.; Meloni, D.; Anfossi, A.; Chessa, B.; Dei Giudici, S.; Graham, S.P.; Oggiano, A.; et al. Comparative phenotypic and functional analyses of the effects of il-10 or tgf-β on porcine macrophages. Animals 2021, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef]
- Barrado-Gil, L.; Galindo, I.; Martínez-Alonso, D.; Viedma, S.; Alonso, C. The ubiquitin-proteasome system is required for African swine fever replication. PLoS ONE 2017, 12, e0189741. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef]
- Freitas, F.B.; Frouco, G.; Martins, C.; Ferreira, F. African swine fever virus encodes for an E2-ubiquitin conjugating enzyme that is mono- and di-ubiquitinated and required for viral replication cycle. Sci. Rep. 2018, 8, 3471. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Li, J.; Guo, S.; Chen, Q.; Zhang, Y.; Liu, Z.; Tan, C.; Chen, H.; Wang, X. African Swine Fever Virus pI215L Inhibits Type I Interferon Signaling by Targeting Interferon Regulatory Factor 9 for Autophagic Degradation. J. Virol. 2022, 96, e00944-22. [Google Scholar] [CrossRef]
- Li, D.; Yang, W.; Li, L.; Li, P.; Ma, Z.; Zhang, J.; Qi, X.; Ren, J.; Ru, Y.; Niu, Q.; et al. African Swine Fever Virus MGF-505-7R Negatively Regulates cGAS–STING-Mediated Signaling Pathway. J. Immunol. 2021, 206, 1844–1857. [Google Scholar] [CrossRef]
- Li, D.; Jing, Z.; Wenping, Y.; Pan, L.; Yi, R.; Weifang, K.; LuLu, L.; Yong, R.; Haixue, Z. African swine fever virus protein MGF-505-7R promotes virulence and pathogenesis by inhibiting JAK1- And JAK2-mediated signaling. J. Biol. Chem. 2021, 297, 101190. [Google Scholar] [CrossRef]
- Kvansakul, M. Viral infection and apoptosis. Viruses 2017, 9, 356. [Google Scholar] [CrossRef]
- Reddy, C.N.; Sankararamakrishnan, R. Designing BH3-Mimetic Peptide Inhibitors for the Viral Bcl-2 Homologues A179L and BHRF1: Importance of Long-Range Electrostatic Interactions. ACS Omega 2021, 6, 26976–26989. [Google Scholar] [CrossRef]
- Banjara, S.; Shimmon, G.L.; Dixon, L.K.; Netherton, C.L.; Hinds, M.G.; Kvansakul, M. Crystal structure of african swine fever virus a179l with the autophagy regulator beclin. Viruses 2019, 11, 789. [Google Scholar] [CrossRef]
- Zheng, X.; Nie, S.; Feng, W.H. Regulation of antiviral immune response by African swine fever virus (ASFV). Virol. Sin. 2022, 37, 157–167. [Google Scholar] [CrossRef]
- Zhou, Y.; Yue, Y.; Fan, S.; Jia, Q.; Ding, X. Advances in Pathophysiology of Triple-Negative Breast Cancer: The Potential of lncRNAs for Clinical Diagnosis, Treatment, and Prognostic Monitoring. Mol. Biotechnol. 2021, 63, 1093–1102. [Google Scholar] [CrossRef]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit To Dephosphorylate eIF2α and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef]
- Jaing, C.; Rowland, R.R.R.; Allen, J.E.; Certoma, A.; Thissen, J.B.; Bingham, J.; Rowe, B.; White, J.R.; Wynne, J.W.; Johnson, D.; et al. Gene expression analysis of whole blood RNA from pigs infected with low and high pathogenic African swine fever viruses. Sci. Rep. 2017, 7, 10115. [Google Scholar] [CrossRef]
- Netherton, C.L.; Connell, S.; Benfield, C.T.O.; Dixon, L.K. The genetics of life and death: Virus-host interactions underpinning resistance to African swine fever, a viral hemorrhagic disease. Front. Genet. 2019, 10, 00402. [Google Scholar] [CrossRef]
- Blome, S.; Franzke, K.; Beer, M. African swine fever—A review of current knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef]
- Frant, M.; Pejsak, Z. African swine fever (ASF) and ticks. No risk of tick-mediated ASF spread in Poland and Baltic states. J. Vet. Res. 2017, 61, 375–380. [Google Scholar] [CrossRef]
- Onyilagha, C.; Nash, M.; Perez, O.; Goolia, M.; Clavijo, A.; Richt, J.A.; Ambagala, A. Meat exudate for detection of african swine fever virus genomic material and anti-asfv antibodies. Viruses 2021, 13, 1744. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.P.; Hutet, E.; Paboeuf, F.; Duhayon, M.; Boinas, F.; de Leon, A.P.; Filatov, S.; Vial, L.; Potier, M.F. Le Comparative vector competence of the Afrotropical soft tick Ornithodoros moubata and Palearctic species, O. erraticus and O. verrucosus, for African swine fever virus strains circulating in Eurasia. PLoS ONE 2019, 14, e0225657. [Google Scholar] [CrossRef]
- Manzano-Romn, R.; Daz-Martn, V.; la de Fuente, J.; Prez-Snchez, R. Soft Ticks as Pathogen Vectors: Distribution, Surveillance and Control. In Parasitology; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Schnettler, E.; Tykalová, H.; Watson, M.; Sharma, M.; Sterken, M.G.; Obbard, D.J.; Lewis, S.H.; McFarlane, M.; Bell-Sakyi, L.; Barry, G.; et al. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res. 2014, 42, 9436–9446. [Google Scholar] [CrossRef]
- Ru, C.; Fazakerley, J.K.; Fragkoudis, R. Antiviral responses of arthropod vectors: An update on recent advances. Virusdisease 2014, 25, 249–260. [Google Scholar] [CrossRef]
- Ali, A.; Zeb, I.; Alouffi, A.; Zahid, H.; Almutairi, M.M.; Ayed Alshammari, F.; Alrouji, M.; Termignoni, C.; Vaz, I.d.S.; Tanaka, T. Host Immune Responses to Salivary Components—A Critical Facet of Tick-Host Interactions. Front. Cell. Infect. Microbiol. 2022, 12, 809052. [Google Scholar] [CrossRef] [PubMed]
- Wasala, N.B.; Jaworski, D.C. Dermacentor variabilis: Characterization and modeling of macrophage migration inhibitory factor with phylogenetic comparisons to other ticks, insects and parasitic nematodes. Exp. Parasitol. 2012, 130, 232–238. [Google Scholar] [CrossRef]
- Feng, W.; Zhou, L.; Zhao, P.; Du, H.; Diao, C.; Zhang, Y.; Liu, J. Genome Assemblies of the Warthog and Kenyan Domestic Pig Provide Insights into Suidae Evolution and Candidate Genes for African Swine Fever Tolerance. bioRxiv 2021. [Google Scholar]
- Palgrave, C.J.; Gilmour, L.; Lowden, C.S.; Lillico, S.G.; Mellencamp, M.A.; Whitelaw, C.B.A. Species-Specific Variation in RELA Underlies Differences in NF-κB Activity: A Potential Role in African Swine Fever Pathogenesis. J. Virol. 2011, 85, 6008–6014. [Google Scholar] [CrossRef]
- McCleary, S.; Strong, R.; McCarthy, R.R.; Edwards, J.C.; Howes, E.L.; Stevens, L.M.; Sánchez-Cordón, P.J.; Núñez, A.; Watson, S.; Mileham, A.J.; et al. Substitution of warthog NF-κB motifs into RELA of domestic pigs is not sufficient to confer resilience to African swine fever virus. Sci. Rep. 2020, 10, 8951. [Google Scholar] [CrossRef]
- Xu, K.; Zhou, Y.; Mu, Y.; Liu, Z.; Hou, S.; Xiong, Y.; Fang, L.; Ge, C.; Wei, Y.; Zhang, X.; et al. CD163 and pAPN double-knockout pigs are resistant to PRRSV and TGEV and exhibit decreased susceptibility to PDCoV while maintaining normal production performance. eLife 2020, 9, 57132. [Google Scholar] [CrossRef]
- Xie, Z.; Pang, D.; Yuan, H.; Jiao, H.; Lu, C.; Wang, K.; Yang, Q.; Li, M.; Chen, X.; Yu, T.; et al. Genetically modified pigs are protected from classical swine fever virus. PLoS Pathog. 2018, 14, 1007193. [Google Scholar] [CrossRef]
- Burkard, C.; Opriessnig, T.; Mileham, A.J.; Stadejek, T.; Ait-Ali, T.; Lillico, S.G.; Whitelaw, C.B.A.; Archibald, A.L. Pigs Lacking the Scavenger Receptor Cysteine-Rich Domain 5 of CD163 Are Resistant to Porcine Reproductive and Respiratory Syndrome Virus 1 Infection. J. Virol. 2018, 92, e00415-18. [Google Scholar] [CrossRef]
- Whitworth, K.M.; Rowland, R.R.R.; Ewen, C.L.; Trible, B.R.; Kerrigan, M.A.; Cino-Ozuna, A.G.; Samuel, M.S.; Lightner, J.E.; McLaren, D.G.; Mileham, A.J.; et al. Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus. Nat. Biotechnol. 2016, 34, 6–8. [Google Scholar] [CrossRef]
- Sánchez-Torres, C.; Gómez-Puertas, P.; Gómez-Del-Moral, M.; Alonso, F.; Escribano, J.M.; Ezquerra, A.; Domínguez, J. Expression of porcine CD163 on monocytes/macrophages correlates with permissiveness to African swine fever infection. Arch. Virol. 2003, 148, 2307–2323. [Google Scholar] [CrossRef]
- Popescu, L.; Gaudreault, N.N.; Whitworth, K.M.; Murgia, M.V.; Nietfeld, J.C.; Mileham, A.; Samuel, M.; Wells, K.D.; Prather, R.S.; Rowland, R.R.R. Genetically edited pigs lacking CD163 show no resistance following infection with the African swine fever virus isolate, Georgia 2007/1. Virology 2017, 501, 102–106. [Google Scholar] [CrossRef]
- Tu, X.; Chu, T.T.; Jeltema, D.; Abbott, K.; Yang, K.; Xing, C.; Han, J.; Dobbs, N.; Yan, N. Interruption of post-Golgi STING trafficking activates tonic interferon signaling. Nat. Commun. 2022, 13, 6977. [Google Scholar] [CrossRef]
- Ding, S.; Diep, J.; Feng, N.; Ren, L.; Li, B.; Ooi, Y.S.; Wang, X.; Brulois, K.F.; Yasukawa, L.L.; Li, X.; et al. STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 2018, 9, 1485. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Viral Protein | Functions | Pathway | References |
|---|---|---|---|
| pC129R | Target Cyclic GMP- AMP To Inhibit the cGAS-STING Signaling Pathway | cGAS-STING | [13] |
| pEP364R | Target Cyclic GMP- AMP To Inhibit the cGAS-STING Signaling Pathway | cGAS-STING | [13] |
| pA137R | Inhibited the nuclear import of IRF3 | cGAS-STING/Ubiquitination | [14] |
| pM1249L | Suppress phosphorylation of TBK1 and degrading IRF3 | cGAS-STING | [15] |
| p17 | Inhibits cGAS-STING signaling pathway through interacting with STING | cGAS-STING | [16] |
| pMGF 505-7R | Inhibit the translocation of IRF3 to the nucleus | cGAS-STING/Ubiquitination | [11] |
| pMGF 360-11L | Inhibits IL-1β, IL-6, and IFN-β secretion | cGAS-STING | [17] |
| p14.5 | Blocks IRF3 phosphorylation | cGAS-STING | [18] |
| MGF 505-11R | Binds to STING and promotes its degradation through the lysosomal and autophagy mechanisms | cGAS-STING | [19] |
| pD345L | Suppress NF-κB signaling by hindering the activity of the IKK kinase | NF-κB | [20] |
| pI226R | Suppresses the stimulation of NF-κB | NF-κB | [21] |
| pF317L | Bound with IκB kinase β (IKKβ) and hindered its phosphorylation, | NF-κB | [22] |
| pA528R | InterruptNF-κB, inhibit downstream promoters, phosphorylation of NF-κB p65, | NF-κB | [23] |
| pI215L | encode the ubiquitin-conjugating enzyme making | Ubiquitination | [24] |
| pE199L | Promotes cell autophagy through the interaction of PYCR2 | Ubiquitination | [25] |
| pK205R | Activates autophagy and the NF-κB signaling pathway | Ubiquitination | [26] |
| pA179L | Anti-apoptotic agent | Apoptosis | [27,28,29] |
| pA224L | Blocks the activation of caspase-3 and enhances the ability of cells to survive | Apoptosis | [30] |
| pEP153R | Prevent apoptosis via activating the p53 and caspase 3 pathways | Apoptosis | [31,32] |
| pDP71L | Dephosphorylation of eIF2α and deactivation of the proapoptotic CHOP factor | Apoptosis | [33] |
| pA238L | Inhibitor of NF-κB pathway | NF-κB | [34] |
| p54 | Enhances apoptosis induction | Apoptosis | [35] |
| pE248R | Inhibit the expression of STING protein | cGAS-STING | [36] |
| pS273R | Interfere with the interaction between IKK and STING. | NF-κB | [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afe, A.E.; Shen, Z.-J.; Guo, X.; Zhou, R.; Li, K. African Swine Fever Virus Interaction with Host Innate Immune Factors. Viruses 2023, 15, 1220. https://doi.org/10.3390/v15061220
Afe AE, Shen Z-J, Guo X, Zhou R, Li K. African Swine Fever Virus Interaction with Host Innate Immune Factors. Viruses. 2023; 15(6):1220. https://doi.org/10.3390/v15061220
Chicago/Turabian StyleAfe, Ayoola Ebenezer, Zhao-Ji Shen, Xiaorong Guo, Rong Zhou, and Kui Li. 2023. "African Swine Fever Virus Interaction with Host Innate Immune Factors" Viruses 15, no. 6: 1220. https://doi.org/10.3390/v15061220
APA StyleAfe, A. E., Shen, Z. -J., Guo, X., Zhou, R., & Li, K. (2023). African Swine Fever Virus Interaction with Host Innate Immune Factors. Viruses, 15(6), 1220. https://doi.org/10.3390/v15061220