
Modeling Drug Resistance Emergence and Transmission in HIV-1 in the UK

 ,
,  on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network
on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Subtyping and Alignment
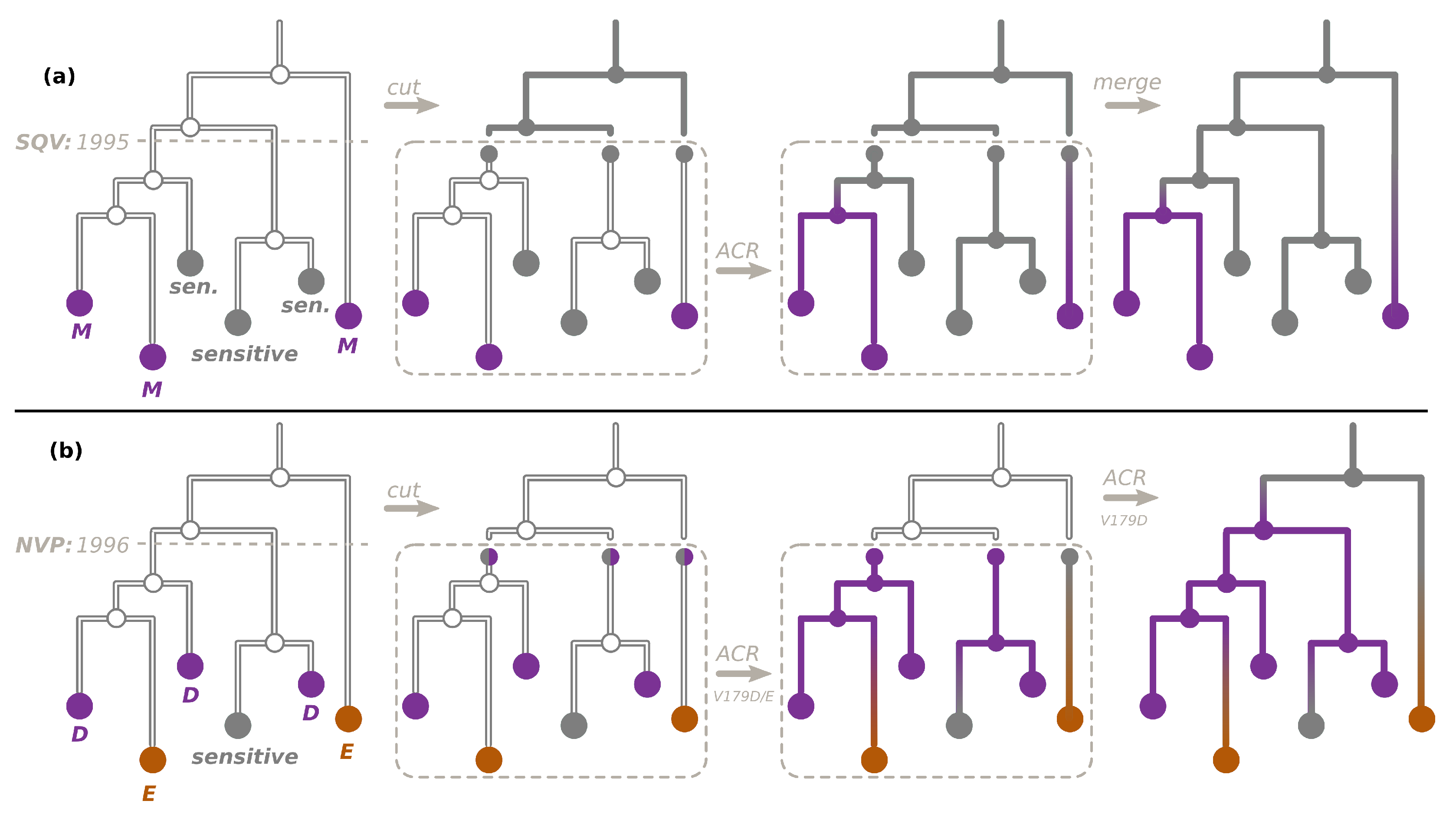
2.2. Transmission Tree Reconstruction
2.3. Ancestral Character Reconstruction
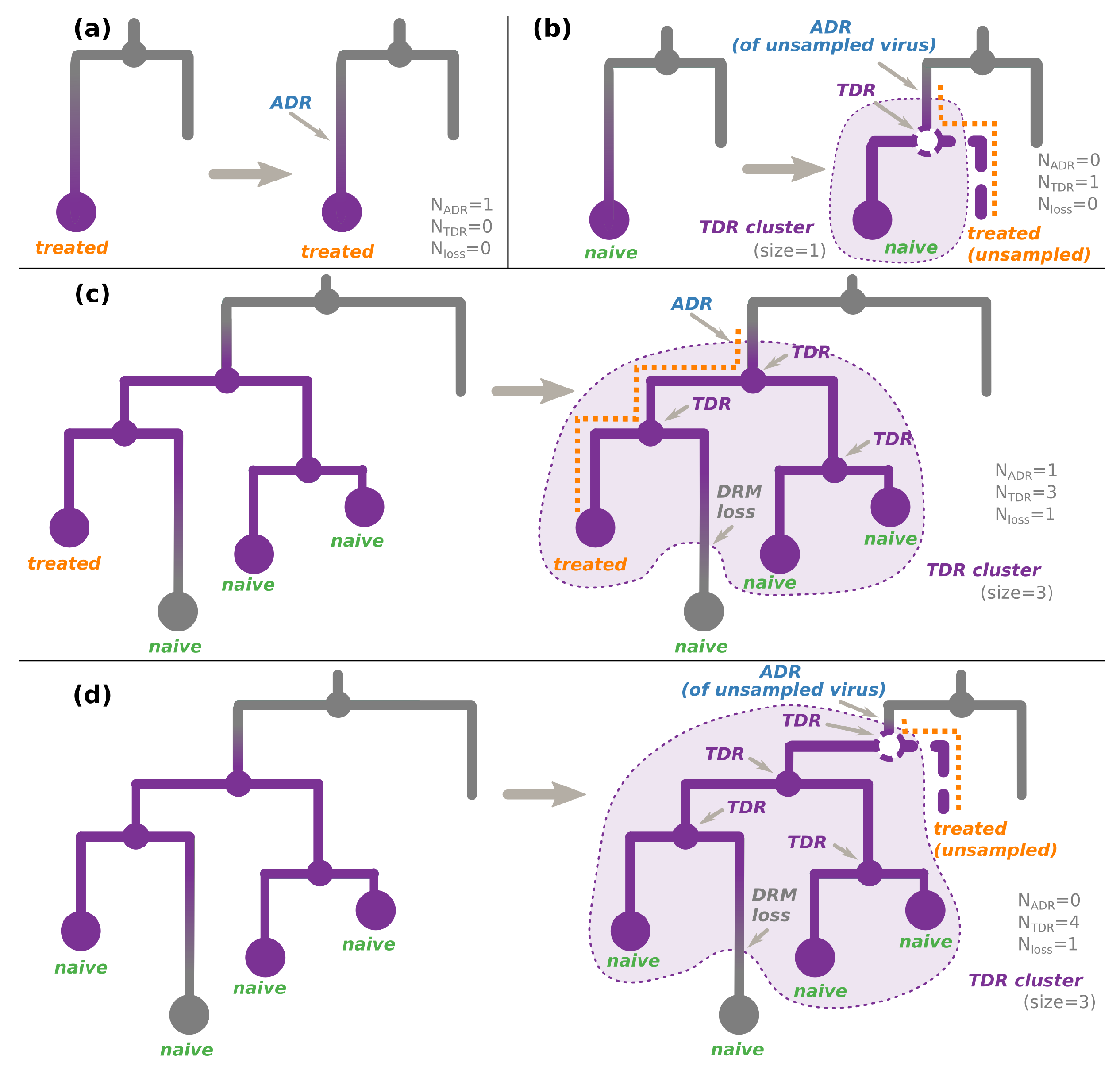
2.4. Transmitted versus Acquired Drug Resistance
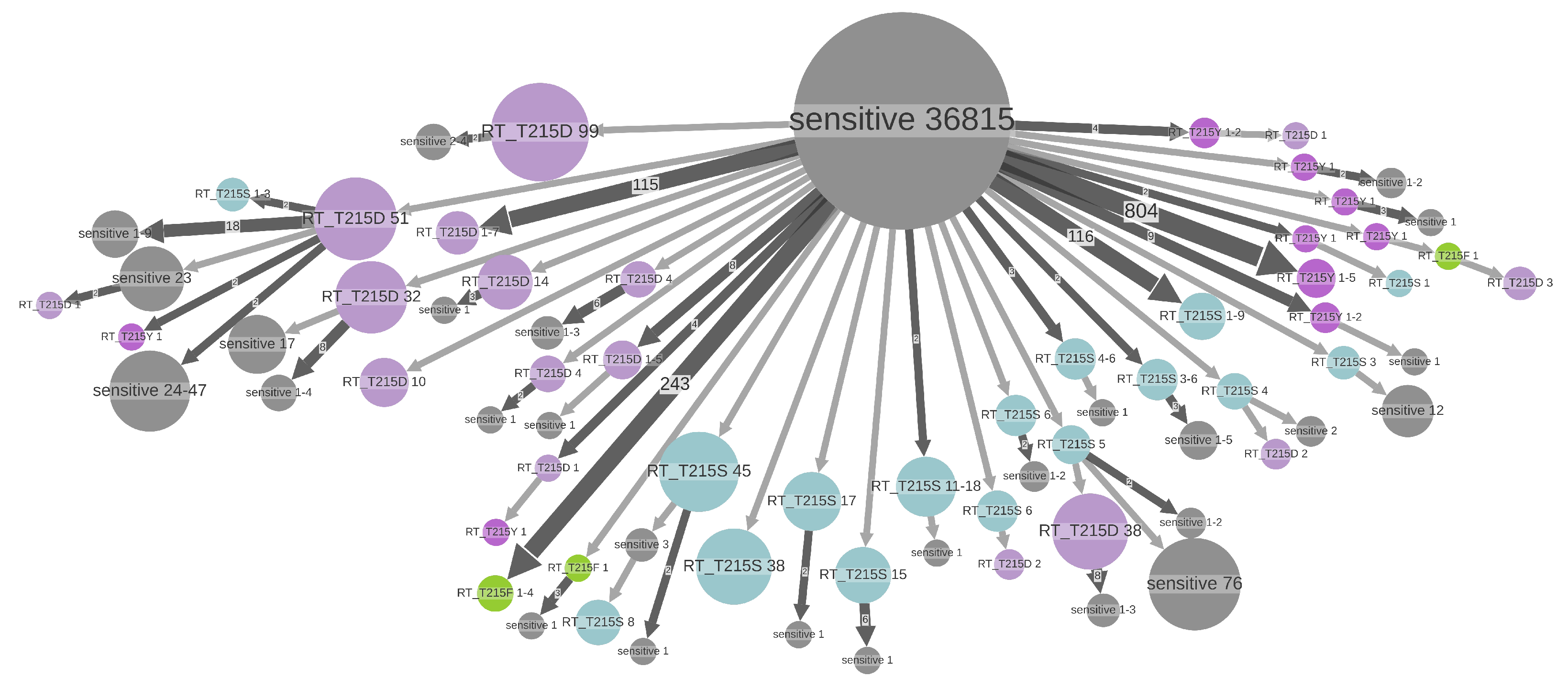
- An internal node whose state was estimated as resistant (i.e., containing the DRM of interest, see Figure 2c,d). As the internal nodes of the tree roughly correspond to transmissions, such a node indicates a transmission of a resistant virus.
- (For non-polymorphic mutations only) A hidden internal node between a node whose DRM status is resistant and its parent node whose DRM status is sensitive, if all the tips in the node’s subtree are treatment-naive. According to the treatment status and the fact that the mutation is non-polymorphic, the initial resistance could not be acquired through treatment pressure, and hence must have been transmitted from a patient who was not sampled (and does not appear in the tree, see Figure 2b,d).
2.4.1. For Non-Polymorphic DRMs
- For treatment-naive tips, the source of their DRM status is:
- For treatment-experienced tips, the source of their DRM status is:
- ADR (+DRM loss if the tip is sensitive) for one of the treatment-experienced tips connected to a TDR cluster (see Figure 2c). The patient corresponding to this tip is assumed to be the source of the TDR cluster. The later DRM loss is possible if the treatment was changed to drugs that do not provoke the DRM in question. For other treated tips connected to this cluster, we assume that they received a resistant virus via TDR. Assuming their treatment was such that it could not provoke the DRM in question, they could later lose it (hence, +DRM loss if they are sensitive);
- ADR for a resistant tip not connected to a TDR cluster (Figure 2a);
- Transmission of a virus without the DRM if the above cases do not apply.
- For the tips whose treatment status is unknown, we consider both cases (naive or resistant) with equal probabilities ().
2.4.2. For Polymorphic DRMs
- ADR for a resistant tip not connected to a TDR cluster (as in Figure 2a, independently of the treatment status);
- ADR (+DRM loss if the tip is sensitive) for one of the tips connected to a TDR cluster (as in Figure 2c, independently of the treatment status). The individual corresponding to this tip is assumed to be the source of the TDR cluster. For other tips connected to this cluster, we assume that they received a resistant virus via TDR. They could later lose it (hence, +DRM loss if they are sensitive);
- Transmission of a virus without the DRM if the above cases do not apply.
2.5. Times of DRM Loss
3. Results
3.1. HIV in the UK
3.2. UK HIV Dataset
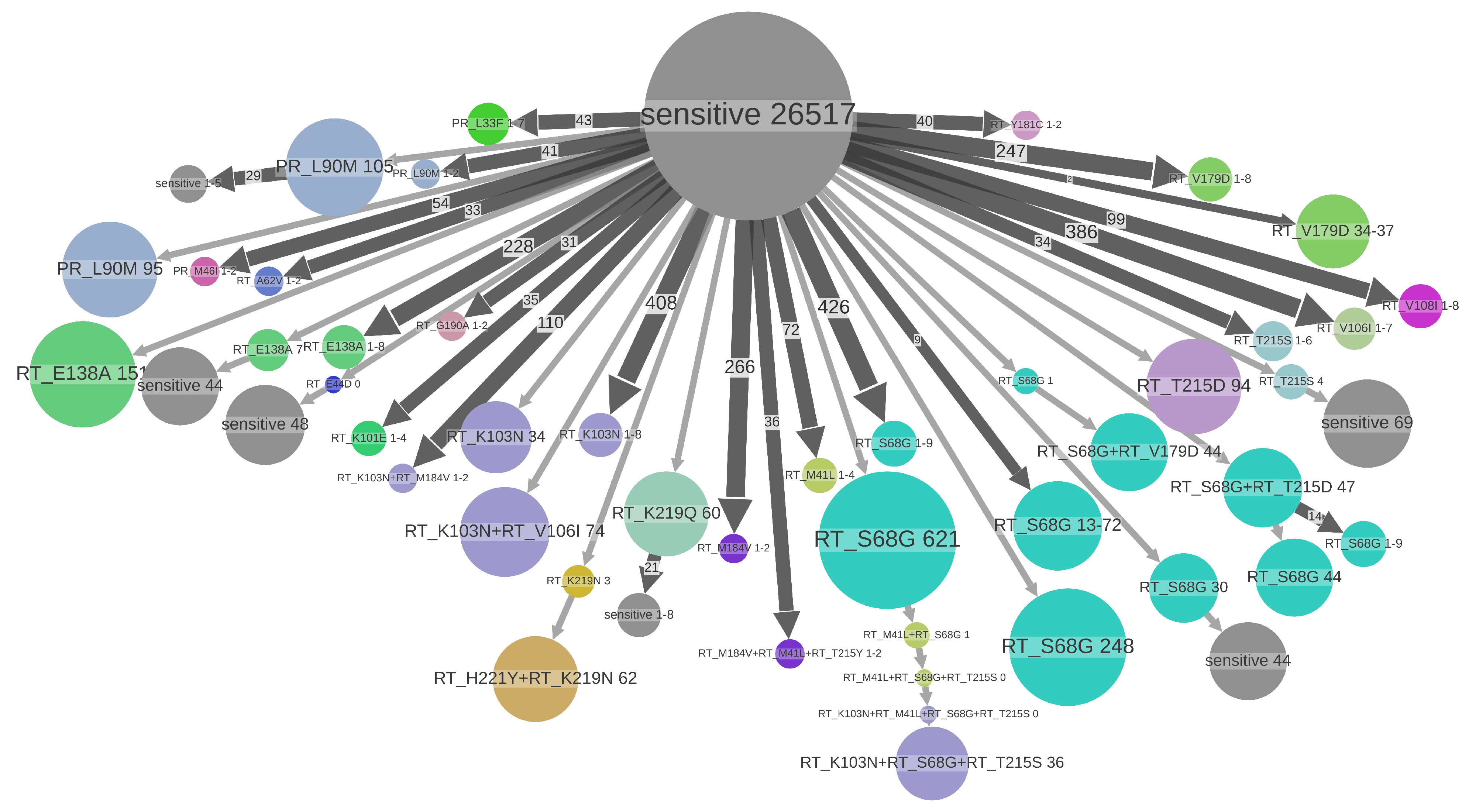
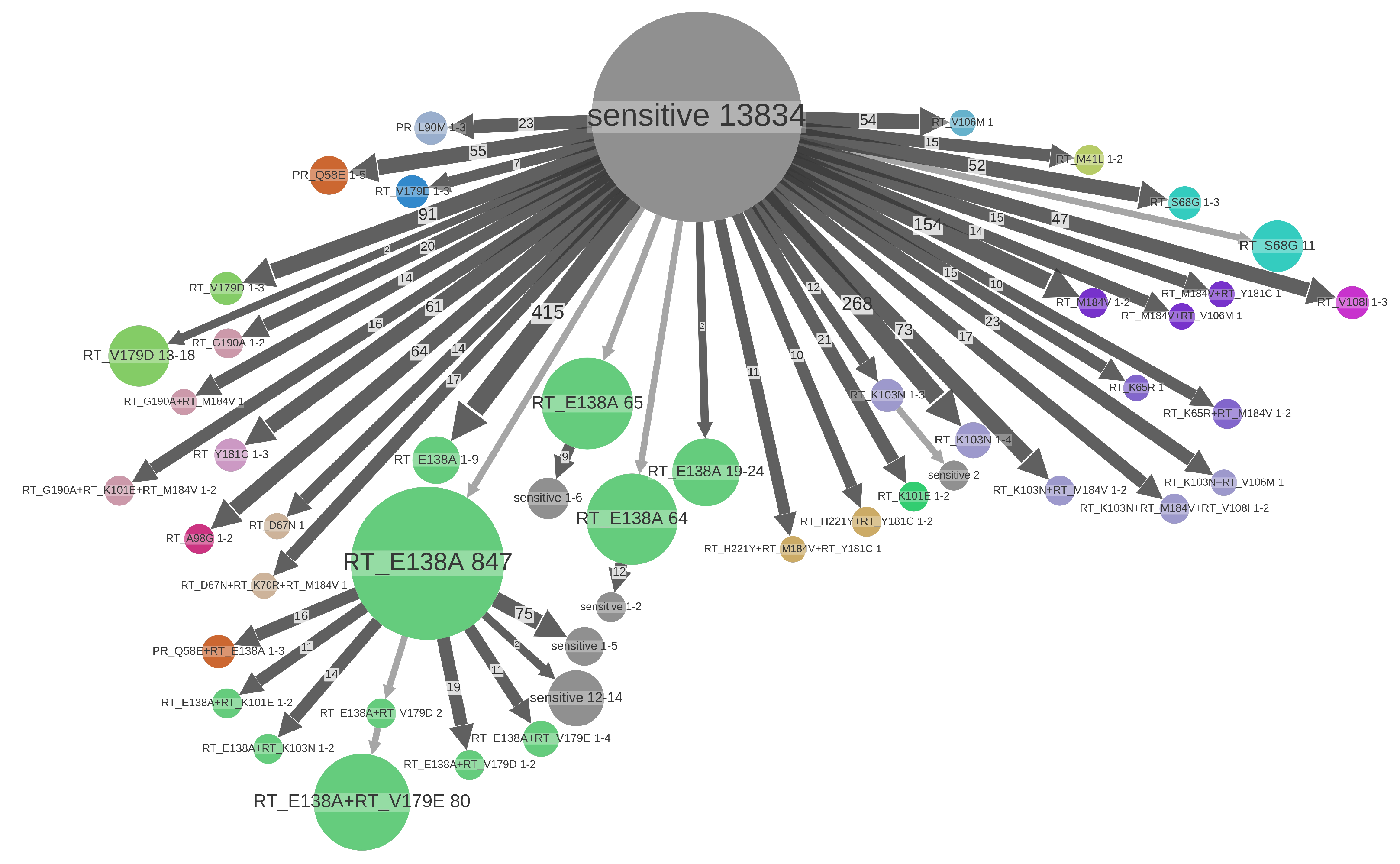
3.3. Drug Resistance Analyses
3.4. DRM Loss Times
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | acquired drug resistance |
| ART | antiretroviral therapy |
| ARV | antiretroviral |
| AZT | zidovudine |
| CI | confidence interval |
| DDI | didanosine |
| DRM | drug resistance mutation |
| ETR | etravirine |
| MAP | maximum a posteriori |
| NFV | nelfinavir |
| NNRTI | non-nucleoside reverse transcriptase inhibitor |
| NRTI | nucleoside reverse transcriptase inhibitor |
| NVP | nevirapine |
| np DRM | non-polymorphic drug resistance mutation |
| PI | protease inhibitor |
| p DRM | polymorphic drug resistance mutation |
| PR | protease |
| RT | reverse transcriptase |
| SQV | saquinavir |
| TDF | tenofovir |
| TDR | transmitted drug resistance |
Appendix A. Analysis Pipelines
Appendix B
| Algorithm A1: Algorithm for Counting , , in a Tree |
 |
Appendix C. Additional Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Number of Cases (% of All) | |||
|---|---|---|---|---|
| of | B | C | ||
| DRMs | All | Non-Polymorphic | All | Non-Polymorphic |
| 0 | 26,859 (68.59%) | 31,518 (80.49%) | 13,661 (72.63%) | 15,795 (83.98%) |
| 1 | 7257 (18.53%) | 3852 (9.84%) | 3174 (16.87%) | 1496 (7.95%) |
| 2 | 2128 (5.43%) | 1243 (3.17%) | 785 (4.17%) | 466 (2.48%) |
| 3 | 852 (2.18%) | 688 (1.76%) | 397 (2.11%) | 362 (1.92%) |
| 4 | 537 (1.37%) | 471 (1.20%) | 258 (1.37%) | 244 (1.30%) |
| 5 | 386 (0.99%) | 350 (0.89%) | 201 (1.07%) | 174 (0.93%) |
| 6 | 308 (0.79%) | 288 (0.74%) | 128 (0.68%) | 113 (0.60%) |
| 7 | 183 (0.47%) | 178 (0.45%) | 80 (0.43%) | 57 (0.30%) |
| 8 | 174 (0.44%) | 163 (0.42%) | 44 (0.23%) | 36 (0.19%) |
| 9 | 121 (0.31%) | 109 (0.28%) | 24 (0.13%) | 21 (0.11%) |
| 10 | 95 (0.24%) | 70 (0.18%) | 19 (0.10%) | 16 (0.09%) |
| 11 | 65 (0.17%) | 70 (0.18%) | 12 (0.06%) | 10 (0.05%) |
| 12 | 50 (0.13%) | 41 (0.10%) | 8 (0.04%) | 5 (0.03%) |
| 13 | 43 (0.11%) | 36 (0.09%) | 6 (0.03%) | 5 (0.03%) |
| 14 | 23 (0.06%) | 25 (0.06%) | 6 (0.03%) | 3 (0.02%) |
| 15 | 23 (0.06%) | 22 (0.06%) | 2 (0.01%) | 2 (0.01%) |
| 16 | 18 (0.05%) | 11 (0.03%) | 0 (0.00%) | 0 (0.00%) |
| 17 | 12 (0.03%) | 14 (0.04%) | 1 (0.01%) | 1 (0.01%) |
| 18 | 10 (0.03%) | 5 (0.01%) | 0 (0.00%) | 0 (0.00%) |
| 19 | 4 (0.01%) | 2 (0.01%) | 0 (0.00%) | 1 (0.01%) |
| 20 | 5 (0.01%) | 2 (0.01%) | 2 (0.01%) | 1 (0.01%) |
| 21 | 5 (0.01%) | 1 (0.00%) | 1 (0.01%) | 1 (0.01%) |
| 22 | 1 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| Date | Total | DRM | Resistant Cases | TDR | ADR | Loss | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Samples | (% of All) | Treatment- | Cases | Cluster | Cases | Cases | ||||
| Experienced | Naive | (% of | Num. | Sizes | (% of | (% of | ||||
| (% of Resistant) | Resistant) | Resistant) | Resistant) | |||||||
| D | 462 (1.2%) | 86 (18.6%) | 334 (72.3%) | 459.25 (99.4%) | 103 | 1–99 | 71.75 (15.5%) | 69 (14.9%) | ||
| F | 257 (0.7%) | 215 (83.7%) | 19 (7.4%) | 41.25 (16.1%) | 37 | 1–4 | 222.75 (86.7%) | 7 (2.7%) | ||
| 14-03-16 | 39159 | S | 378 (1.0%) | 59 (15.6%) | 293 (77.5%) | 364.25 (96.4%) | 115 | 1–45 | 51.75 (13.7%) | 38 (10.1%) |
| Y | 883 (2.3%) | 790 (89.5%) | 37 (4.2%) | 119.50 (13.5%) | 102.5 | 1–5 | 785.50 (89.0%) | 22 (2.5%) | ||
| D | 441 (1.2%) | 83 (18.8%) | 322 (73.0%) | 437.25 (99.1%) | 102 | 1–88 | 70.75 (16.0%) | 67 (15.2%) | ||
| F | 256 (0.7%) | 214 (83.6%) | 19 (7.4%) | 41.25 (16.1%) | 37 | 1–4 | 221.75 (86.6%) | 7 (2.7%) | ||
| 17-12-14 | 36258 | S | 358 (1.0%) | 53 (14.8%) | 284 (79.3%) | 348.75 (97.4%) | 109 | 1–44 | 47.25 (13.2%) | 38 (10.6%) |
| Y | 880 (2.4%) | 788 (89.5%) | 37 (4.2%) | 118.00 (13.4%) | 102 | 1–5 | 783.00 (89.0%) | 21 (2.4%) | ||
| D | 314 (1.4%) | 61 (19.4%) | 234 (74.5%) | 309.50 (98.6%) | 83 | 1–47 | 60.50 (19.3%) | 56 (17.8%) | ||
| F | 241 (1.1%) | 204 (84.6%) | 17 (7.1%) | 36.50 (15.1%) | 33.5 | 1–4 | 209.50 (86.9%) | 5 (2.1%) | ||
| 17-12-09 | 22540 | S | 226 (1.0%) | 34 (15.0%) | 178 (78.8%) | 222.00 (98.2%) | 75 | 1–21 | 35.00 (15.5%) | 31 (13.7%) |
| Y | 837 (3.7%) | 752 (89.8%) | 34 (4.1%) | 99.00 (11.8%) | 87 | 1–5 | 751.00 (89.7%) | 13 (1.6%) | ||
| D | 123 (1.6%) | 31 (25.2%) | 85 (69.1%) | 109.50 (89.0%) | 45.5 | 1–21 | 37.50 (30.5%) | 24 (19.5%) | ||
| F | 185 (2.5%) | 160 (86.5%) | 14 (7.6%) | 24.00 (13.0%) | 24 | 1–2 | 163.00 (88.1%) | 2 (1.1%) | ||
| 17-12-04 | 7511 | S | 70 (0.9%) | 17 (24.3%) | 43 (61.4%) | 63.75 (91.1%) | 34 | 1–8 | 22.25 (31.8%) | 16 (22.9%) |
| Y | 655 (8.7%) | 597 (91.1%) | 24 (3.7%) | 54.25 (8.3%) | 51 | 1–4 | 602.75 (92.0%) | 2 (0.3%) | ||
| D | 28 (1.8%) | 9 (32.1%) | 17 (60.7%) | 20.00 (71.4%) | 14.5 | 1–2 | 10.00 (35.7%) | 2 (7.1%) | ||
| F | 42 (2.7%) | 41 (97.6%) | 1 (2.4%) | 2.00 (4.8%) | 2 | 1–2 | 40.00 (95.2%) | |||
| 17-12-99 | 1576 | S | 9 (0.6%) | 3 (33.3%) | 4 (44.4%) | 5.00 (55.6%) | 4.5 | 1–2 | 4.00 (44.4%) | |
| Y | 205 (13.0%) | 187 (91.2%) | 6 (2.9%) | 12.25 (6.0%) | 12 | 1–2 | 192.75 (94.0%) | |||
| D | 1 (14.3%) | 1 (100.0%) | 1.00 (100.0%) | |||||||
| F | 1 (14.3%) | 1 (100.0%) | 1.00 (100.0%) | |||||||
| 14-11-96 | 7 | S | ||||||||
| Y | 2 (28.6%) | 2 (100.0%) | 2.00 (100.0%) | |||||||
| DRM | Class | Num. Data Points B | Num. Data Points C | ||||
|---|---|---|---|---|---|---|---|
| Left- | Right- | Interval- | Left- | Right- | Interval- | ||
| Censored | Censored | ||||||
| PR:L33F | PI | 36 | 17 | 0 | |||
| PR:M46I | PI | 95 | 8 | 1 | |||
| PR:I54V | PI | 77 | 7 | 4 | |||
| PR:V82A | PI | 80 | 17 | 1 | |||
| PR:L90M | PI | 153 | 35 | 0 | |||
| RT:M41L | NRTI | 238 | 68 | 4 | |||
| RT:E44D | NRTI | 50 | 9 | 1 | |||
| RT:A62V | NRTI | 68 | 14 | 0 | |||
| RT:D67N | NRTI | 231 | 25 | 4 | |||
| RT:K70R | NRTI | 216 | 4 | 2 | |||
| RT:K103N | NNRTI | 612 | 90 | 8 | 310 | 11 | 5 |
| RT:V108I | NNRTI | 148 | 9 | 2 | |||
| RT:Y181C | NNRTI | 264 | 9 | 5 | |||
| RT:M184V | NRTI | 825 | 7 | 3 | 408 | 4 | 7 |
| RT:G190A | NNRTI | 185 | 14 | 4 | |||
| RT:L210W | NRTI | 140 | 19 | 0 | |||
| RT:T215D | NRTI | 24 | 43 | 2 | |||
| RT:T215F | NRTI | 69 | 3 | 2 | |||
| RT:T215S | NRTI | 30 | 35 | 3 | |||
| RT:T215Y | NRTI | 223 | 5 | 1 | |||
| RT:K219E | NRTI | 72 | 8 | 1 | |||
| RT:K219Q | NRTI | 79 | 32 | 3 | |||
| RT:K219N | NRTI | 38 | 18 | 1 | |||
| RT:H221Y | NNRTI | 153 | 17 | 1 | |||
References
- Larder, B.A.; Kemp, S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 1989, 246, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Lepri, A.C.; Sabin, C.A.; Staszewski, S.; Hertogs, K.; Müller, A.; Rabenau, H.; Phillips, A.N.; Miller, V. Resistance Profiles in Patients with Viral Rebound on Potent Antiretroviral Therapy. J. Infect. Dis. 2000, 181, 1143–1147. [Google Scholar] [CrossRef]
- Hué, S.; Gifford, R.J.; Dunn, D.; Fernhill, E.; Pillay, D.; UK Collaborative Group on HIV Drug Resistance. Demonstration of sustained drug-resistant human immunodeficiency virus type 1 lineages circulating among treatment-naïve individuals. J. Virol. 2009, 83, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Mourad, R.; Chevennet, F.; Dunn, D.T.; Fearnhill, E.; Delpech, V.; Asboe, D.; Gascuel, O.; Hue, S.; UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network. A phylotype-based analysis highlights the role of drug-naive HIV-positive individuals in the transmission of antiretroviral resistance in the UK. AIDS 2015, 29, 1917–1925. [Google Scholar] [CrossRef]
- Castro, H.; Pillay, D.; Cane, P.; Asboe, D.; Cambiano, V.; Phillips, A.; Dunn, D.T.; for the UK Collaborative Group on HIV Drug Resistance; Aitken, C.; Asboe, D.; et al. Persistence of HIV-1 transmitted drug resistance mutations. J. Infect. Dis. 2013, 208, 1459–1463. [Google Scholar] [CrossRef]
- The World Health Organization. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing Hiv Infection: Recommendations for a Public Health Approach, 2nd ed.; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Stadler, T.; Bonhoeffer, S. Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120198. [Google Scholar] [CrossRef]
- Stadler, T.; Kühnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef]
- Kühnert, D.; Kouyos, R.; Shirreff, G.; Pečerska, J.; Scherrer, A.U.; Böni, J.; Yerly, S.; Klimkait, T.; Aubert, V.; Günthard, H.F.; et al. Quantifying the fitness cost of HIV-1 drug resistance mutations through phylodynamics. PLoS Pathog. 2018, 14, e1006895. [Google Scholar] [CrossRef]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-Dörner, L.; Bonsall, D.; Hoppe, A.; Brown, A.L.; de Oliveira, T.; et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef]
- Fitch, W.M. Toward Defining the Course of Evolution: Minimum Change for a Specific Tree Topology. Syst. Biol. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Kühnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Phylodynamics with Migration: A Computational Framework to Quantify Population Structure from Genomic Data. Mol. Biol. Evol. 2016, 33, 2102–2116. [Google Scholar] [CrossRef]
- Ishikawa, S.A.; Zhukova, A.; Iwasaki, W.; Gascuel, O. A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios. Mol. Biol. Evol. 2019, 36, 2069–2085. [Google Scholar] [CrossRef]
- Dunn, D.; Pillay, D. UK HIV drug resistance database: Background and recent outputs. J. HIV Ther. 2007, 12, 97–98. [Google Scholar]
- Kuiken, C.; Korber, B.; Shafer, R.W. HIV sequence databases. AIDS Rev. 2003, 5, 52–61. [Google Scholar]
- Schultz, A.K.; Zhang, M.; Leitner, T.; Kuiken, C.; Korber, B.; Morgenstern, B.; Stanke, M. A jumping profile Hidden Markov Model and applications to recombination sites in HIV and HCV genomes. BMC Bioinform. 2006, 7, 265. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Bennett, D.E.; Camacho, R.J.; Otelea, D.; Kuritzkes, D.R.; Fleury, H.; Kiuchi, M.; Heneine, W.; Kantor, R.; Jordan, M.R.; Schapiro, J.M.; et al. Drug Resistance Mutations for Surveillance of Transmitted HIV-1 Drug-Resistance: 2009 Update. PLoS ONE 2009, 4, e4724. [Google Scholar] [CrossRef]
- Castoe, T.A.; de Koning, A.P.J.; Kim, H.M.; Gu, W.; Noonan, B.P.; Naylor, G.; Jiang, Z.J.; Parkinson, C.L.; Pollock, D.D. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8986–8991. [Google Scholar] [CrossRef]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef]
- To, T.H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Shafer, R.W.; Jung, D.R.; Betts, B.J. Human immunodeficiency virus type 1 reverse transcriptase and protease mutation search engine for queries. Nat. Med. 2000, 6, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef]
- Zhukova, A.; Voznica, J.; Felipe, M.D.; To, T.H.; Pérez, L.; Martínez, Y.; Pintos, Y.; Méndez, M.; Gascuel, O.; Kouri, V. Cuban history of CRF19 recombinant subtype of HIV-1. PLoS Pathog. 2021, 17, e1009786. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, L.; Vella, S. A brief history of antiretroviral therapy of HIV infection: Success and challenges. Annali dell’Istituto Superiore di Sanita 2011, 47, 44–48. [Google Scholar] [PubMed]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J.; Feinberg, J.E.; Balfour, H.H.; Deyton, L.R.; et al. A Controlled Trial of Two Nucleoside Analogues plus Indinavir in Persons with Human Immunodeficiency Virus Infection and CD4 Cell Counts of 200 per Cubic Millimeter or Less. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar] [CrossRef]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with Indinavir, Zidovudine, and Lamivudine in Adults with Human Immunodeficiency Virus Infection and Prior Antiretroviral Therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 Infection with Early Antiretroviral Therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef]
- Rodger, A.J.; Cambiano, V.; Bruun, T.; Vernazza, P.; Collins, S.; van Lunzen, J.; Corbelli, G.M.; Estrada, V.; Geretti, A.M.; Beloukas, A.; et al. Sexual Activity Without Condoms and Risk of HIV Transmission in Serodifferent Couples When the HIV-Positive Partner Is Using Suppressive Antiretroviral Therapy. JAMA 2016, 316, 171–181. [Google Scholar] [CrossRef]
- Blassel, L.; Tostevin, A.; Villabona-Arenas, C.J.; Peeters, M.; Hué, S.; Gascuel, O.; on behalf of the UK HIV Drug Resistance Database. Using machine learning and big data to explore the drug resistance landscape in HIV. PLoS Comput. Biol. 2021, 17, e1008873. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Fourment, M.; Kosakovsky Pond, S.L. Inconsistencies in Estimating the Age of HIV-1 Subtypes Due to Heterotachy. Mol. Biol. Evol. 2012, 29, 451–456. [Google Scholar] [CrossRef]
- Bletsa, M.; Suchard, M.A.; Ji, X.; Gryseels, S.; Vrancken, B.; Baele, G.; Worobey, M.; Lemey, P. Divergence dating using mixed effects clock modelling: An application to HIV-1. Virus Evol. 2019, 5, vez036. [Google Scholar] [CrossRef]
- Goudsmit, J.; de Ronde, A.; de Rooij, E.; de Boer, R. Broad spectrum of in vivo fitness of human immunodeficiency virus type 1 subpopulations differing at reverse transcriptase codons 41 and 215. J. Virol. 1997, 71, 4479–4484. [Google Scholar] [CrossRef]
- Tambuyzer, L.; Azijn, H.; Rimsky, L.T.; Vingerhoets, J.; Lecocq, P.; Kraus, G.; Picchio, G.; de Béthune, M.P. Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antivir. Ther. 2009, 14, 103–109. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Wang, X.; Han, J.; Jia, L.; Li, T.; Li, J.; Li, L. Natural presence of V179E and rising prevalence of E138G in HIV-1 reverse transcriptase in CRF55_01B viruses. Infect. Genet. Evol. 2020, 77, 104098. [Google Scholar] [CrossRef]
- Scherrer, A.U.; von Wyl, V.; Yang, W.L.; Kouyos, R.D.; Böni, J.; Yerly, S.; Klimkait, T.; Aubert, V.; Cavassini, M.; Battegay, M.; et al. Emergence of Acquired HIV-1 Drug Resistance Almost Stopped in Switzerland: A 15-Year Prospective Cohort Analysis. Clin. Infect. Dis. 2016, 62, 1310–1317. [Google Scholar] [CrossRef]
- Rossetti, B.; Di Giambenedetto, S.; Torti, C.; Postorino, M.C.; Punzi, G.; Saladini, F.; Gennari, W.; Borghi, V.; Monno, L.; Pignataro, A.R.; et al. Evolution of transmitted HIV-1 drug resistance and viral subtypes circulation in Italy from 2006 to 2016. HIV Med. 2018, 19, 619–628. [Google Scholar] [CrossRef]
- Pingarilho, M.; Pimentel, V.; Diogo, I.; Fernandes, S.; Miranda, M.; Pineda-Pena, A.; Libin, P.; Theys, K.; Martins, M.R.O.; Vandamme, A.M.; et al. Increasing Prevalence of HIV-1 Transmitted Drug Resistance in Portugal: Implications for First Line Treatment Recommendations. Viruses 2020, 12, 1238. [Google Scholar] [CrossRef]
- Villabona-Arenas, C.J.; Vidal, N.; Guichet, E.; Serrano, L.; Delaporte, E.; Gascuel, O.; Peeters, M. In-depth analysis of HIV-1 drug resistance mutations in HIV-infected individuals failing first-line regimens in West and Central Africa. AIDS 2016, 30, 2577–2589. [Google Scholar] [CrossRef]
- Abela, I.A.; Scherrer, A.U.; Böni, J.; Yerly, S.; Klimkait, T.; Perreau, M.; Hirsch, H.H.; Furrer, H.; Calmy, A.; Schmid, P.; et al. Emergence of Drug Resistance in the Swiss HIV Cohort Study Under Potent Antiretroviral Therapy Is Observed in Socially Disadvantaged Patients. Clin. Infect. Dis. 2020, 70, 297–303. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]
- Lemoine, F.; Gascuel, O. Gotree/Goalign: Toolkit and Go API to facilitate the development of phylogenetic workflows. NAR Genom. Bioinform. 2021, 3, lqab075. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]





| DRM | Class | 1st ARV | Resistant Cases | TDR | ADR | Loss | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| and | (% of All) | Treatment- | Cases | Cluster | Cases | Cases | ||||
| Its Date | Experienced | Naive | (% of | Num. | Sizes | (% of | (% of | |||
| (% of Resistant) | Resistant) | Resistant) | Resistant) | |||||||
| RT:S68G | NRTI | polym. | 3178 (8.1%) | 436 (13.7%) | 2482 (78.1%) | 2436.00 (76.7%) | 249 | 1–759 | 803.00 (25.3%) | 61 (1.9%) |
| RT:K103N | NNRTI | NVP’96 | 2025 (5.2%) | 1104 (54.5%) | 745 (36.8%) | 1071.51 (52.9%) | 516.5 | 1–78 | 1088.49 (53.8%) | 135 (6.7%) |
| RT:M184V | NRTI | AZT’87 | 1899 (4.8%) | 1642 (86.5%) | 110 (5.8%) | 343.62 (18.1%) | 278.5 | 1–4 | 1667.38 (87.8%) | 112 (5.9%) |
| RT:M41L | NRTI | AZT’87 | 1513 (3.9%) | 982 (64.9%) | 428 (28.3%) | 618.50 (40.9%) | 305.5 | 1–55 | 968.50 (64.0%) | 74 (4.9%) |
| RT:V106I | NNRTI | polym. | 1051 (2.7%) | 217 (20.6%) | 715 (68.0%) | 540.00 (51.4%) | 150 | 1–74 | 647.00 (61.6%) | 136 (12.9%) |
| RT:D67N | NRTI | AZT’87 | 1035 (2.6%) | 806 (77.9%) | 150 (14.5%) | 273.00 (26.4%) | 170.5 | 1–21 | 794.00 (76.7%) | 32 (3.1%) |
| RT:T215Y | NRTI | AZT’87 | 883 (2.3%) | 790 (89.5%) | 37 (4.2%) | 119.50 (13.5%) | 102.5 | 1–5 | 785.50 (89.0%) | 22 (2.5%) |
| RT:E138A | NNRTI | polym. | 862 (2.2%) | 163 (18.9%) | 637 (73.9%) | 523.00 (60.7%) | 117 | 1–158 | 393.00 (45.6%) | 54 (6.3%) |
| PR:L90M | PI | SQV’95 | 849 (2.2%) | 480 (56.5%) | 289 (34.0%) | 460.77 (54.3%) | 128 | 1–114 | 450.23 (53.0%) | 62 (7.3%) |
| RT:V179D | NNRTI | polym. | 790 (2.0%) | 151 (19.1%) | 559 (70.8%) | 415.00 (52.5%) | 93 | 1–45 | 438.00 (55.4%) | 63 (8.0%) |
| RT:K70R | NRTI | AZT’87 | 711 (1.8%) | 610 (85.8%) | 54 (7.6%) | 143.75 (20.2%) | 98.5 | 1–7 | 615.25 (86.5%) | 48 (6.8%) |
| RT:L210W | NRTI | AZT’87 | 705 (1.8%) | 520 (73.8%) | 140 (19.9%) | 205.00 (29.1%) | 147 | 1–9 | 524.00 (74.3%) | 24 (3.4%) |
| RT:Y181C | NNRTI | NVP’96 | 694 (1.8%) | 495 (71.3%) | 115 (16.6%) | 208.00 (30.0%) | 148 | 1–12 | 509.00 (73.3%) | 23 (3.3%) |
| RT:K219Q | NRTI | AZT’87 | 563 (1.4%) | 322 (57.2%) | 194 (34.5%) | 307.25 (54.6%) | 99 | 1–92 | 303.75 (54.0%) | 48 (8.5%) |
| RT:H221Y | NNRTI | NVP’96 | 475 (1.2%) | 269 (56.6%) | 162 (34.1%) | 220.00 (46.3%) | 87 | 1–64 | 267.00 (56.2%) | 12 (2.5%) |
| RT:T215D | NRTI | AZT’87 | 462 (1.2%) | 86 (18.6%) | 334 (72.3%) | 459.25 (99.4%) | 103 | 1–99 | 71.75 (15.5%) | 69 (14.9%) |
| RT:G190A | NNRTI | NVP’96 | 447 (1.1%) | 342 (76.5%) | 68 (15.2%) | 117.25 (26.2%) | 97 | 1–6 | 350.75 (78.5%) | 21 (4.7%) |
| RT:V108I | NNRTI | NVP’96 | 429 (1.1%) | 219 (51.0%) | 167 (38.9%) | 230.00 (53.6%) | 166 | 1–8 | 232.00 (54.1%) | 33 (7.7%) |
| PR:M46I | PI | SQV’95 | 378 (1.0%) | 246 (65.1%) | 97 (25.7%) | 140.39 (37.1%) | 108.5 | 1–6 | 250.61 (66.3%) | 13 (3.4%) |
| RT:T215S | NRTI | AZT’87 | 378 (1.0%) | 59 (15.6%) | 293 (77.5%) | 364.25 (96.4%) | 115 | 1–45 | 51.75 (13.7%) | 38 (10.1%) |
| PR:V82A | PI | SQV’95 | 295 (0.8%) | 216 (73.2%) | 51 (17.3%) | 88.02 (29.8%) | 53 | 1–11 | 218.98 (74.2%) | 12 (4.1%) |
| RT:E44D | NRTI | AZT’87 | 294 (0.8%) | 180 (61.2%) | 93 (31.6%) | 129.62 (44.1%) | 77 | 1–29 | 183.38 (62.4%) | 19 (6.5%) |
| RT:K101E | NNRTI | NVP’96 | 276 (0.7%) | 189 (68.5%) | 64 (23.2%) | 94.50 (34.2%) | 71.5 | 1–9 | 190.50 (69.0%) | 9 (3.3%) |
| RT:K219E | NRTI | AZT’87 | 262 (0.7%) | 192 (73.3%) | 43 (16.4%) | 74.75 (28.5%) | 51.5 | 1–9 | 192.25 (73.4%) | 5 (1.9%) |
| RT:T215F | NRTI | AZT’87 | 257 (0.7%) | 215 (83.7%) | 19 (7.4%) | 41.25 (16.1%) | 37 | 1–4 | 222.75 (86.7%) | 7 (2.7%) |
| RT:A62V | NRTI | AZT’87 | 251 (0.6%) | 147 (58.6%) | 81 (32.3%) | 114.50 (45.6%) | 58.5 | 1–27 | 147.50 (58.8%) | 11 (4.4%) |
| PR:I54V | PI | SQV’95 | 243 (0.6%) | 182 (74.9%) | 33 (13.6%) | 62.52 (25.7%) | 43 | 1–6 | 191.48 (78.8%) | 11 (4.5%) |
| RT:L74V | NRTI | DDI’91 | 242 (0.6%) | 200 (82.6%) | 17 (7.0%) | 38.25 (15.8%) | 36 | 1–3 | 207.75 (85.8%) | 4 (1.7%) |
| RT:K219N | NRTI | AZT’87 | 238 (0.6%) | 92 (38.7%) | 127 (53.4%) | 161.00 (67.6%) | 23 | 1–113 | 81.00 (34.0%) | 4 (1.7%) |
| PR:L33F | PI | SQV’95 | 230 (0.6%) | 117 (50.9%) | 92 (40.0%) | 126.12 (54.8%) | 70 | 1–10 | 114.88 (49.9%) | 11 (4.8%) |
| RT:K65R | NRTI | AZT’87 | 225 (0.6%) | 170 (75.6%) | 19 (8.4%) | 50.88 (22.6%) | 42 | 1–2 | 187.12 (83.2%) | 13 (5.8%) |
| DRM | Class | 1st ARV | Resistant Cases | TDR | ADR | Loss | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| and | (% of All) | Treatment- | Cases | Cluster | Cases | Cases | ||||
| Its Date | Experienced | Naive | (% of | Num. | Sizes | (% of | (% of | |||
| (% of Resistant) | Resistant) | Resistant) | Resistant) | |||||||
| RT:E138A | NNRTI | polym. | 2176 (11.6%) | 512 (23.5%) | 1381 (63.5%) | 1802.00 (82.8%) | 136 | 2–1178 | 531.00 (24.4%) | 157 (7.2%) |
| RT:M184V | NRTI | AZT’87 | 1009 (5.4%) | 789 (78.2%) | 79 (7.8%) | 213.88 (21.2%) | 197 | 1–4 | 833.12 (82.6%) | 38 (3.8%) |
| RT:K103N | NNRTI | NVP’96 | 882 (4.7%) | 605 (68.6%) | 182 (20.6%) | 317.12 (36.0%) | 267 | 1–5 | 615.88 (69.8%) | 51 (5.8%) |
| RT:Y181C | NNRTI | NVP’96 | 419 (2.2%) | 299 (71.4%) | 56 (13.4%) | 108.38 (25.9%) | 98 | 1–4 | 321.62 (76.8%) | 11 (2.6%) |
| RT:V106M | NNRTI | NVP’96 | 381 (2.0%) | 301 (79.0%) | 36 (9.4%) | 71.25 (18.7%) | 66 | 1–4 | 319.75 (83.9%) | 10 (2.6%) |
| RT:V179D | NNRTI | polym. | 294 (1.6%) | 99 (33.7%) | 159 (54.1%) | 105.00 (35.7%) | 39 | 1–19 | 212.00 (72.1%) | 23 (7.8%) |
| RT:D67N | NRTI | AZT’87 | 289 (1.5%) | 215 (74.4%) | 25 (8.7%) | 65.25 (22.6%) | 56.5 | 1–4 | 228.75 (79.2%) | 5 (1.7%) |
| RT:G190A | NNRTI | NVP’96 | 287 (1.5%) | 213 (74.2%) | 34 (11.8%) | 71.25 (24.8%) | 65.5 | 1–4 | 224.75 (78.3%) | 9 (3.1%) |
| RT:K65R | NRTI | AZT’87 | 244 (1.3%) | 199 (81.6%) | 15 (6.1%) | 38.50 (15.8%) | 36.5 | 1–2 | 211.50 (86.7%) | 6 (2.5%) |
| RT:K101E | NNRTI | NVP’96 | 244 (1.3%) | 164 (67.2%) | 54 (22.1%) | 82.75 (33.9%) | 73.5 | 1–4 | 168.25 (69.0%) | 7 (2.9%) |
| RT:A98G | NNRTI | NVP’96 | 239 (1.3%) | 115 (48.1%) | 78 (32.6%) | 112.12 (46.9%) | 99 | 1–4 | 126.88 (53.1%) | |
| RT:K70R | NRTI | AZT’87 | 196 (1.0%) | 152 (77.6%) | 19 (9.7%) | 38.62 (19.7%) | 35.5 | 1–4 | 161.38 (82.3%) | 4 (2.0%) |
| RT:V108I | NNRTI | NVP’96 | 194 (1.0%) | 114 (58.8%) | 55 (28.4%) | 77.75 (40.1%) | 72 | 1–3 | 123.25 (63.5%) | 7 (3.6%) |
| RT:H221Y | NNRTI | NVP’96 | 173 (0.9%) | 123 (71.1%) | 27 (15.6%) | 45.75 (26.4%) | 42.5 | 1–2 | 133.25 (77.0%) | 6 (3.5%) |
| RT:M41L | NRTI | AZT’87 | 171 (0.9%) | 117 (68.4%) | 25 (14.6%) | 51.72 (30.2%) | 43.5 | 1–5 | 120.28 (70.3%) | 1 (0.6%) |
| RT:S68G | NRTI | polym. | 160 (0.9%) | 51 (31.9%) | 87 (54.4%) | 37.00 (23.1%) | 18 | 2–12 | 124.00 (77.5%) | 1 (0.6%) |
| PR:Q58E | PI | polym. | 153 (0.8%) | 31 (20.3%) | 97 (63.4%) | 49.00 (32.0%) | 24 | 2–12 | 106.00 (69.3%) | 2 (1.3%) |
| RT:T215Y | NRTI | AZT’87 | 137 (0.7%) | 97 (70.8%) | 13 (9.5%) | 37.97 (27.7%) | 31.5 | 1–5 | 105.03 (76.7%) | 6 (4.4%) |
| RT:V179E | NNRTI | NVP’96 | 120 (0.6%) | 11 (9.2%) | 34 (28.3%) | 108.25 (90.2%) | 24 | 1–80 | 12.75 (10.6%) | 1 (0.8%) |
| RT:K219E | NRTI | AZT’87 | 109 (0.6%) | 80 (73.4%) | 15 (13.8%) | 25.50 (23.4%) | 24.5 | 1–3 | 83.50 (76.6%) | |
| PR:L90M | PI | SQV’95 | 108 (0.6%) | 62 (57.4%) | 22 (20.4%) | 43.00 (39.8%) | 35.5 | 1–4 | 67.00 (62.0%) | 2 (1.9%) |
| B | C | ||
|---|---|---|---|
| total | 58,569 | 27,151 | |
| filtered by patient (first only, % of total) | 40,055 (68%) | 19,139 (70%) | |
| – without temporal outliers (% of filtered) | 39,159 (99%) | 18,809 (98%) | |
| – with DRM(s) (% of w/o outliers) | 12,300 (31%) | 5148 (27%) | |
| – w. 1 DRM (% of w/o outliers) | 7257 (19%) | 3174 (17%) | |
| – w. DRMs (% of w/o outliers) | 5043 (13%) | 1974 (10%) | |
| – with np DRM(s) (% of w/o outliers) | 7641 (20%) | 3014 (16%) | |
| – w. 1 np DRM (% of w/o outliers) | 3852 (10%) | 1496 (8%) | |
| – w. np DRMs (% of w/o outliers) | 3789 (10%) | 1518 (8%) | |
| – with p DRM(s) (% of w/o outliers) | 5740 (15%) | 2673 (14%) | |
| – w. 1 p DRM(s) (% of w/o outliers) | 5416 (14%) | 2538 (13%) | |
| – w. p DRM(s) (% of w/o outliers) | 324 (1%) | 135 (1%) | |
| Number | treatment-naive (% of w/o outliers) | 28,175 (72%) | 12,286 (65%) |
| of | – with DRM(s) (% of tr.-naive) | 7091 (25%) | 2361 (19%) |
| sequences | – with np DRM(s) (% of tr.-naive) | 3364 (12%) | 829 (7%) |
| – with p DRM(s) (% of tr.-naive) | 4260 (15%) | 1656 (13%) | |
| treatment-experienced (% of w/o outliers) | 7732 (20%) | 4503 (24%) | |
| – with DRM(s) (% of tr.-experienced) | 4141 (54%) | 2112 (47%) | |
| – with np DRM(s) (% of tr.-experienced) | 3618 (47%) | 1730 (38%) | |
| – with p DRM(s) (% of tr.-experienced) | 971 (13%) | 665 (15%) | |
| treatment-unknown (% of w/o outliers) | 3252 (8%) | 2020 (11%) | |
| Root date (95% CI) | 1965 (’59–’65) | 1944 (’29–’49) | |
| Mutation rate (95% CI) | 1.9 (1.8–1.9) | 1.4 (1.3–1.4) | |
| Phylogenetic diversity = | 0.014 | 0.019 | |
| DRM | Class | Loss Duration + CI (Years) | ||
|---|---|---|---|---|
| Our Estimate B | Our Estimate C | Castro et al.’13 [5] | ||
| PR:L33F | PI | 3.1 (2.2–4.8) | ||
| PR:M46I | PI | 1.1 (0.7–1.9) | ||
| PR:I54V | PI | 2.2 (1.6–3.6) | 3.3 (1.4–7.8) | |
| PR:V82A | PI | 3.3 (2.4–4.9) | 5.1 (1.8–14.8) | |
| PR:L90M | PI | 2.7 (2.1–3.7) | 5.8 (2.2–15.3) | |
| RT:M41L | NRTI | 4.3 (3.6–5.2) | 8.6 (4.6–16.0) | |
| RT:E44D | NRTI | 3.0 (2.0–5.6) | ||
| RT:A62V | NRTI | 2.4 (1.8–3.6) | ||
| RT:D67N | NRTI | 2.1 (1.7–2.8) | 6.0 (2.1–16.9) | |
| RT:K70R | NRTI | 1.3 (1.1–2.1) | 1.8 (0.8–4.0) | |
| RT:K103N | NNRTI | 2.2 (2.0–2.6) | 1.1 (0.9–1.6) | 3.7 (2.0–6.8) |
| RT:V108I | NNRTI | 1.3 (1.0–1.9) | ||
| RT:Y181C | NNRTI | 1.3 (1.0–2.1) | 3.7 (2.0–6.8) | |
| RT:M184V | NRTI | 0.6 (0.5–0.8) | 0.6 (0.5–0.8) | 1.0 (0.5–2.0) |
| RT:G190A | NNRTI | 1.8 (1.5–2.5) | 3.6 (1.2–15.5) | |
| RT:L210W | NRTI | 2.9 (2.3–4.1) | 4.8 (2.1–11.2) | |
| RT:T215D | NRTI | 9.3 (6.4–12.2) | ||
| RT:T215F | NRTI | 1.8 (1.6–3.1) | 1.2 (0.3–4.6) | |
| RT:T215S | NRTI | 6.8 (4.7–9.6) | ||
| RT:T215Y | NRTI | 1.1 (1.0–1.8) | 1.7 (0.8–3.4) | |
| RT:K219Q | NRTI | 4.9 (3.8–6.4) | 15.8 (3.6–70.0) | |
| RT:K219N | NRTI | 3.7 (2.6–5.7) | 4.6 (1.0–22.4) | |
| RT:K219E | NRTI | 1.7 (1.3–3.0) | ||
| RT:H221Y | NNRTI | 1.7 (1.4–2.5) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhukova, A.; Dunn, D.; Gascuel, O., on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network. Modeling Drug Resistance Emergence and Transmission in HIV-1 in the UK. Viruses 2023, 15, 1244. https://doi.org/10.3390/v15061244
Zhukova A, Dunn D, Gascuel O on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network. Modeling Drug Resistance Emergence and Transmission in HIV-1 in the UK. Viruses. 2023; 15(6):1244. https://doi.org/10.3390/v15061244
Chicago/Turabian StyleZhukova, Anna, David Dunn, and Olivier Gascuel on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network. 2023. "Modeling Drug Resistance Emergence and Transmission in HIV-1 in the UK" Viruses 15, no. 6: 1244. https://doi.org/10.3390/v15061244
APA StyleZhukova, A., Dunn, D., & Gascuel, O., on behalf of the UK HIV Drug Resistance Database & the Collaborative HIV, Anti-HIV Drug Resistance Network. (2023). Modeling Drug Resistance Emergence and Transmission in HIV-1 in the UK. Viruses, 15(6), 1244. https://doi.org/10.3390/v15061244