

Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Purification of Native Human C1q
2.2. Purification of Recombinant ghA, ghB, and ghC Modules of Human C1q
2.3. Purification of Native Human C4BP
2.4. Direct-Binding ELISA
2.5. Cell Culture
2.6. Viral Cell Entry Assay
2.6.1. Preparation of SARS-CoV-2 Pseudotyped Lentiviral Particles
2.6.2. Treatment of SARS-CoV-2 Pseudotyped Lentiviral Particles
2.7. Luciferase Reporter Assay
2.8. NF-κB Activity Assay
2.9. Cell Binding Assay
2.10. Modulation of SARS-CoV-2 Pseudoparticle-Induced Infection by C1q or C4BP
Quantitative qRT-PCR Analysis
2.11. Statistical Analysis
3. Results
3.1. Human C1q and C4BP Bind to SARS-CoV-2 Spike and RBD Proteins
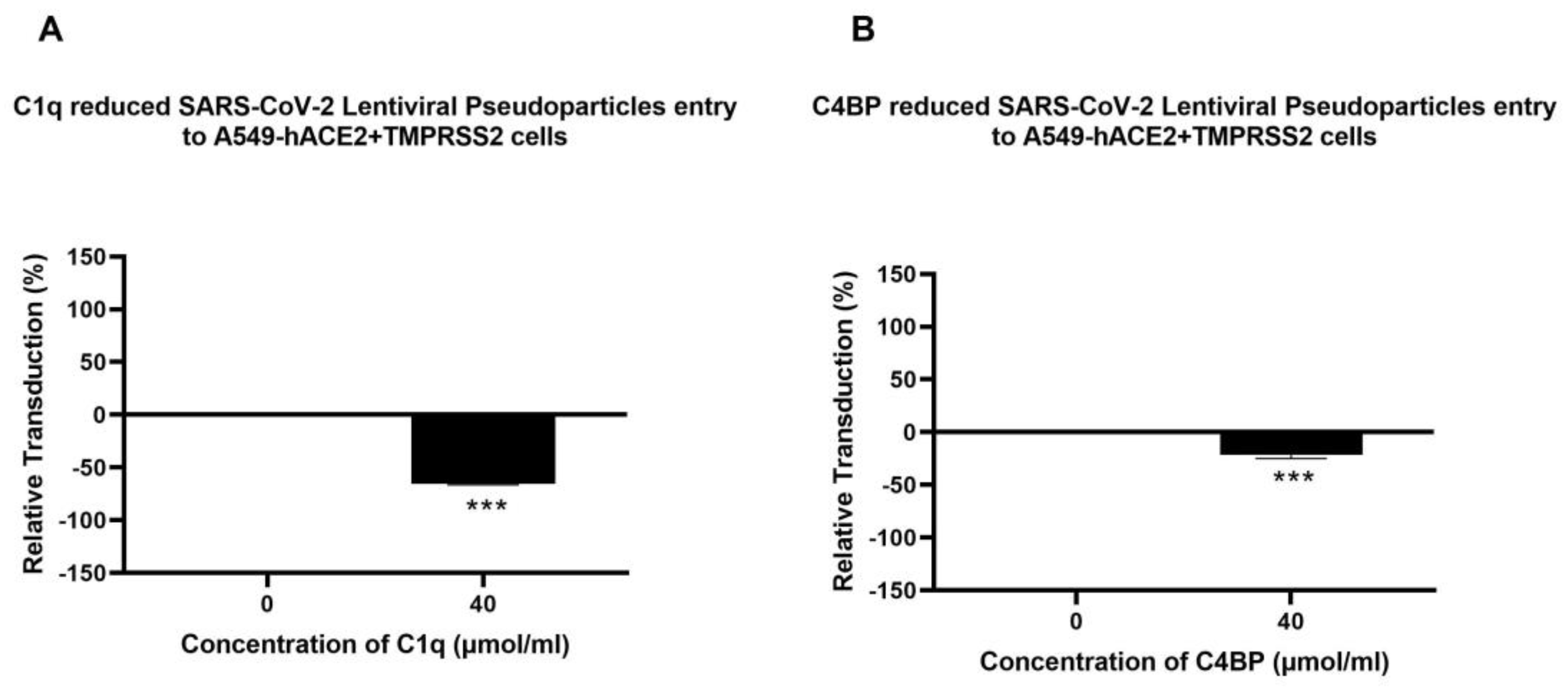
3.2. Human C1q, Recombinant Globular Head Modules, and C4BP Inhibit SARS-CoV-2 Pseudoparticle Transduction
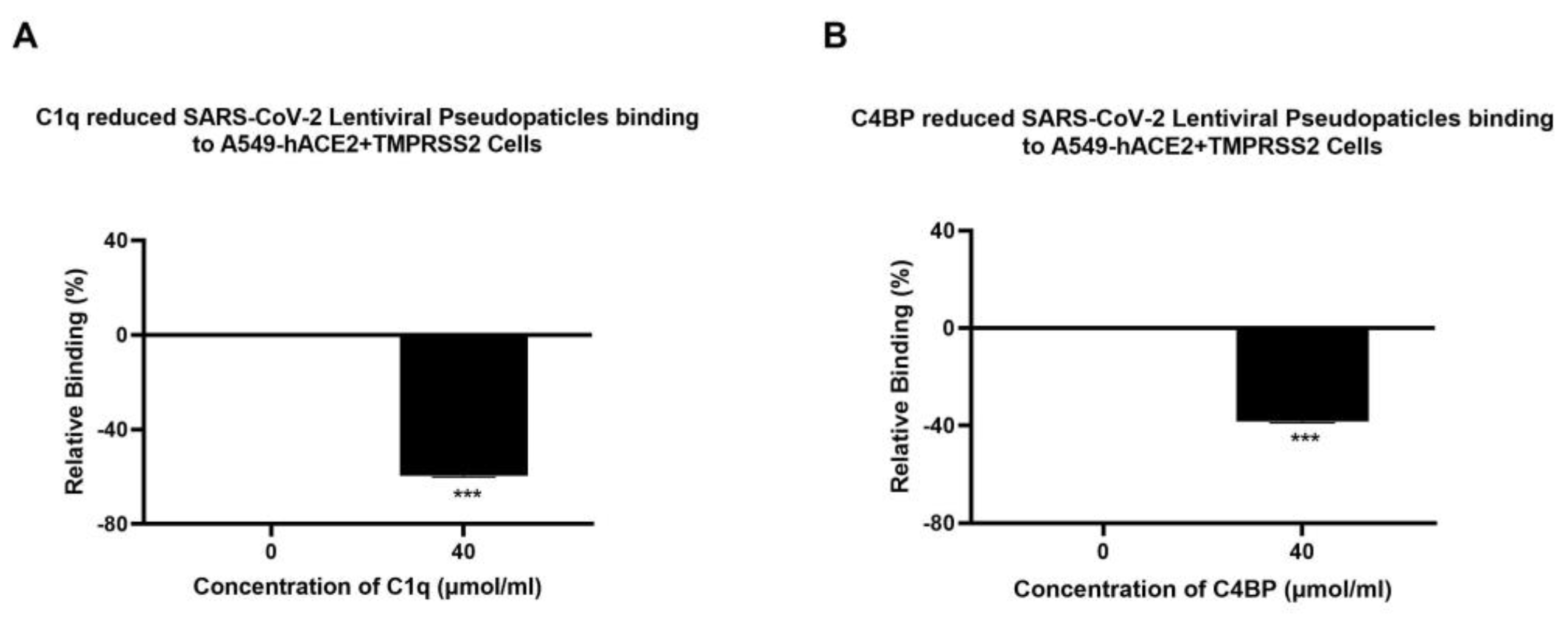
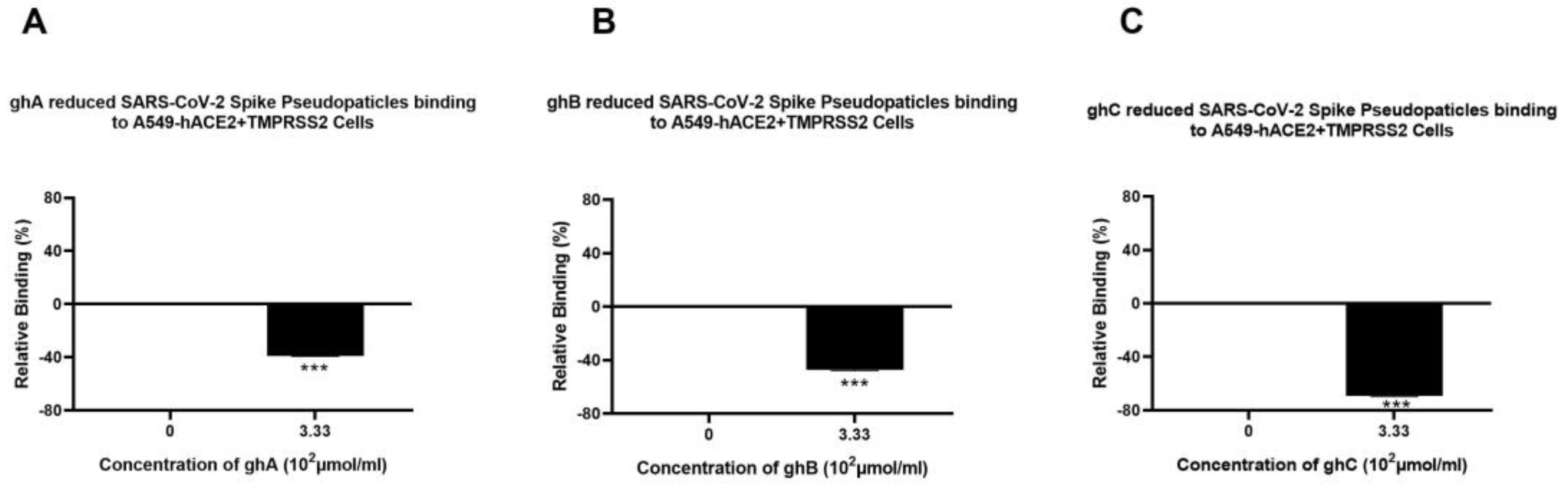
3.3. Human C1q, Recombinant Globular Head Modules, and C4BP Inhibit SARS-CoV-2 Pseudoparticle Binding to ACE2- and TMPRSS2-Expressing A549 Cells
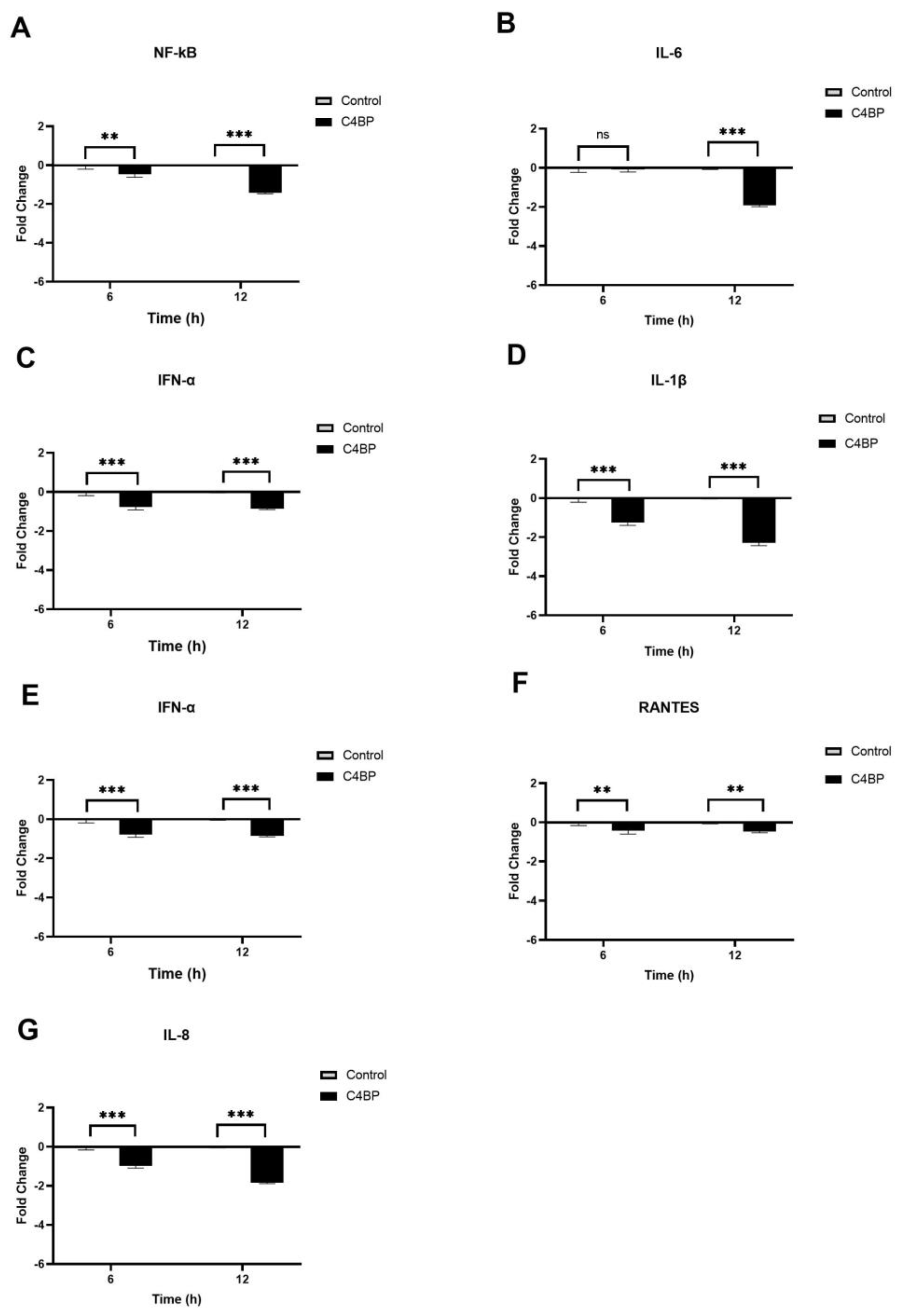
3.4. C1q and C4BP Attenuate Inflammatory Response in SARS-CoV-2 Pseudoparticles Challenged A549-hACE2+TMPRSS2 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, evaluation, and treatment of coronavirus (COVID-19). In StatPearls [Internet]; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific immune response and the pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef] [PubMed]
- Varghese, P.M.; Tsolaki, A.G.; Yasmin, H.; Shastri, A.; Ferluga, J.; Vatish, M.; Madan, T.; Kishore, U. Host-pathogen interaction in COVID-19: Pathogenesis, potential therapeutics and vaccination strategies. Immunobiology 2020, 225, 152008. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I–molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugaiah, V.; Varghese, P.M.; Beirag, N.; DeCordova, S.; Sim, R.B.; Kishore, U. Complement proteins as soluble pattern recognition receptors for pathogenic viruses. Viruses 2021, 13, 824. [Google Scholar] [CrossRef]
- Houser, K.V.; Broadbent, A.J.; Gretebeck, L.; Vogel, L.; Lamirande, E.W.; Sutton, T.; Bock, K.W.; Minai, M.; Orandle, M.; Moore, I.N. Enhanced inflammation in New Zealand white rabbits when MERS-CoV reinfection occurs in the absence of neutralizing antibody. PLoS Pathog. 2017, 13, e1006565. [Google Scholar] [CrossRef] [Green Version]
- Duchateau, J.; Haas, M.; Schreyen, H.; Radoux, L.; Sprangers, I.; Noel, F.; Braun, M.; Lamy, M. Complement activation in patients at risk of developing the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1984, 130, 1058–1064. [Google Scholar]
- Wong, L.-Y.R.; Perlman, S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—Are we our own worst enemy? Nat. Rev. Immunol. 2022, 22, 47–56. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, e140711. [Google Scholar] [CrossRef]
- Alosaimi, B.; Mubarak, A.; Hamed, M.E.; Almutairi, A.Z.; Alrashed, A.A.; AlJuryyan, A.; Enani, M.; Alenzi, F.Q.; Alturaiki, W. Complement anaphylatoxins and inflammatory cytokines as prognostic markers for COVID-19 severity and in-hospital mortality. Front. Immunol. 2021, 12, 668725. [Google Scholar] [CrossRef]
- Siggins, M.K.; Davies, K.; Fellows, R.; Thwaites, R.S.; Baillie, J.K.; Semple, M.G.; Openshaw, P.J.; Zelek, W.M.; Harris, C.L.; Morgan, B.P. Alternative pathway dysregulation in tissues drives sustained complement activation and predicts outcome across the disease course in COVID-19. Immunology 2023, 168, 473–492. [Google Scholar] [CrossRef]
- Mehlhop, E.; Diamond, M.S. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006, 203, 1371–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Erp, E.A.; Luytjes, W.; Ferwerda, G.; Van Kasteren, P.B. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front. Immunol. 2019, 10, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.Q.; Mozdzanowska, K.; Gerhard, W. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. J. Virol. 2002, 76, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef]
- Rodríguez de Córdoba, S.; Sanchez-Corral, P.; Rey-Campos, J. Structure of the gene coding for the alpha polypeptide chain of the human complement component C4b-binding protein. J. Exp. Med. 1991, 173, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Varghese, P.M.; Murugaiah, V.; Beirag, N.; Temperton, N.; Khan, H.A.; Alrokayan, S.H.; Al-Ahdal, M.N.; Nal, B.; Al-Mohanna, F.A.; Sim, R.B.; et al. C4b Binding Protein Acts as an Innate Immune Effector Against Influenza A Virus. Front. Immunol. 2021, 11, 585361. [Google Scholar] [CrossRef]
- Chaudhary, N.; Jayaraman, A.; Reinhardt, C.; Campbell, J.D.; Bosmann, M. A single-cell lung atlas of complement genes identifies the mesothelium and epithelium as prominent sources of extrahepatic complement proteins. Mucosal Immunol. 2022, 15, 927–939. [Google Scholar] [CrossRef]
- Varghese, P.M.; Kishore, U.; Rajkumari, R. Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. Int. J. Mol. Sci. 2022, 23, 3045. [Google Scholar] [CrossRef]
- Tan, L.A.; Yu, B.; Sim, F.C.; Kishore, U.; Sim, R.B. Complement activation by phospholipids: The interplay of factor H and C1q. Protein Cell 2010, 1, 1033–1049. [Google Scholar] [CrossRef] [Green Version]
- Kishore, U.; Gupta, S.K.; Perdikoulis, M.V.; Kojouharova, M.S.; Urban, B.C.; Reid, K.B. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, and C chains. J. Immunol. 2003, 171, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Sim, E.; Sim, R. Enzymic assay of C3b receptor on intact cells and solubilized cells. Biochem. J. 1983, 210, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Genova, C.; Sampson, A.; Scott, S.; Cantoni, D.; Mayora-Neto, M.; Bentley, E.; Mattiuzzo, G.; Wright, E.; Derveni, M.; Auld, B. Production, titration, neutralisation, storage and lyophilisation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lentiviral pseudotypes. Bio-Protocol 2021, 11, e4236. [Google Scholar] [CrossRef] [PubMed]
- Antos, A.; Kwong, M.L.; Balmorez, T.; Villanueva, A.; Murakami, S. Unusually high risks of COVID-19 mortality with age-related comorbidities: An adjusted meta-analysis method to improve the risk assessment of mortality using the comorbid mortality data. Infect. Dis. Rep. 2021, 13, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef]
- Varghese, P.M.; Mukherjee, S.; Al-Mohanna, F.A.; Saleh, S.M.; Almajhdi, F.N.; Beirag, N.; Alkahtani, S.H.; Rajkumari, R.; Nal Rogier, B.; Sim, R.B. Human properdin released by infiltrating neutrophils can modulate Influenza A virus infection. Front. Immunol. 2021, 12, 747654. [Google Scholar] [CrossRef]
- Murugaiah, V.; Tsolaki, A.G.; Kishore, U. Collectins: Innate immune pattern recognition molecules. Adv. Exp. Med. Biol. 2020, 1204, 75–127. [Google Scholar]
- Zissel, G.; Ernst, M.; Rabe, K.; Papadopoulos, T.; Magnussen, H.; Schlaak, M.; Müller-Quernheim, J. Human alveolar epithelial cells type II are capable of regulating T-cell activity. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2000, 48, 66–75. [Google Scholar]
- Chang, C.-W.; Parsi, K.M.; Somasundaran, M.; Vanderleeden, E.; Liu, P.; Cruz, J.; Cousineau, A.; Finberg, R.W.; Kurt-Jones, E.A. A newly engineered A549 cell line expressing ACE2 and TMPRSS2 is highly permissive to SARS-CoV-2, including the delta and omicron variants. Viruses 2022, 14, 1369. [Google Scholar] [CrossRef]
- Cantoni, D.; Mayora-Neto, M.; Temperton, N. The role of pseudotype neutralization assays in understanding SARS CoV-2. Oxf. Open Immunol. 2021, 2, iqab005. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F.; Mazzetti, P.; Pistello, M. Viral infection neutralization tests: A focus on severe acute respiratory syndrome-coronavirus-2 with implications for convalescent plasma therapy. Rev. Med. Virol. 2021, 31, e2170. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, H.; Saha, S.; Butt, M.T.; Modi, R.K.; George, A.J.T.; Kishore, U. SARS-CoV-2: Pathogenic Mechanisms and Host Immune Response. Adv. Exp. Med. Biol. 2021, 1313, 99–134. [Google Scholar] [PubMed]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Q.; Cheng, A.; Wang, Y.; Li, H.; Hu, L.; Zhao, X.; Wang, T.; He, F. Cytokines and their relationship with the severity and prognosis of coronavirus disease 2019 (COVID-19): A retrospective cohort study. BMJ Open 2020, 10, e041471. [Google Scholar] [CrossRef]
- Satyam, A.; Tsokos, M.G.; Brook, O.R.; Hecht, J.L.; Moulton, V.R.; Tsokos, G.C. Activation of classical and alternative complement pathways in the pathogenesis of lung injury in COVID-19. Clin. Immunol. 2021, 226, 108716. [Google Scholar] [CrossRef]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.-D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell 2020, 27, 962–973.e7. [Google Scholar] [CrossRef]
- Chen, L.; Guan, W.-J.; Qiu, Z.-E.; Xu, J.-B.; Bai, X.; Hou, X.-C.; Sun, J.; Qu, S.; Huang, Z.-X.; Lei, T.-L. SARS-CoV-2 nucleocapsid protein triggers hyperinflammation via protein-protein interaction-mediated intracellular Cl− accumulation in respiratory epithelium. Signal Transduct. Target. Ther. 2022, 7, 255. [Google Scholar] [CrossRef]
- Anand, G.; Perry, A.M.; Cummings, C.L.; St Raymond, E.; Clemens, R.A.; Steed, A.L. Surface proteins of SARS-CoV-2 drive airway epithelial cells to induce IFN-dependent inflammation. J. Immunol. 2021, 206, 3000–3009. [Google Scholar] [CrossRef]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. NF-κB pathway as a potential target for treatment of critical stage COVID-19 patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.-L.; Yuen, K.-S.; Castaño-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865. [Google Scholar] [CrossRef] [PubMed]
- Mardi, A.; Meidaninikjeh, S.; Nikfarjam, S.; Majidi Zolbanin, N.; Jafari, R. Interleukin-1 in COVID-19 infection: Immunopathogenesis and possible therapeutic perspective. Viral Immunol. 2021, 34, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.; Ross, R.; Frydas, I.; Kritas, S. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [PubMed]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.D.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Cauchois, R.; Koubi, M.; Delarbre, D.; Manet, C.; Carvelli, J.; Blasco, V.B.; Jean, R.; Fouche, L.; Bornet, C.; Pauly, V. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc. Natl. Acad. Sci. USA 2020, 117, 18951–18953. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.S. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020, 92, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Shin, E.-C. The type I interferon response in COVID-19: Implications for treatment. Nat. Rev. Immunol. 2020, 20, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe 2020, 28, 455–464.e2. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Mantelli, F.; Allegretti, M.; Balk, R.A. The role of interleukin-8 in lung inflammation and injury: Implications for the management of COVID-19 and hyperinflammatory acute respiratory distress syndrome. Front. Pharmacol. 2022, 12, 3931. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, H.; Matthay, M.; Hebert, C.; Broaddus, V. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J. Clin. Investig. 1995, 96, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Thevarajan, I.; Nguyen, T.H.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef] [Green Version]
- Alturaiki, W.H. Evaluation of CC chemokine ligand 5 (CCL5) chemokine, interleukin 5 (IL-5) cytokine, and eosinophil counts as potential biomarkers in Saudi patients with chronic asthma during sandstorms. Cureus 2020, 12, e7809. [Google Scholar]
- Patterson, B.K.; Seethamraju, H.; Dhody, K.; Corley, M.J.; Kazempour, K.; Lalezari, J.; Pang, A.P.; Sugai, C.; Mahyari, E.; Francisco, E.B. CCR5 inhibition in critical COVID-19 patients decreases inflammatory cytokines, increases CD8 T-cells, and decreases SARS-CoV2 RNA in plasma by day 14. Int. J. Infect. Dis. 2021, 103, 25–32. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| 18S | 5′-ATGGCCGTTCTTAGTTGGTG-3′ | 5′-CGCTGAGCCAGTCAGTGTAG-3′ |
| TNF-α | 5′-AGCCCATGTTGTAGCAAACC-3′ | 5′-TGAGGTACAGGCCCTCTGAT-3′ |
| IL-6 | 5′-GAAAGCAGCAAGAGGCACT-3 | 5′-TTTCACCAGGCAAGTCTCCT-3′ |
| IL-8 | 5′-GTGCAGTTTTTGCCAAGGAG-3′ | 5′-CACCCAGTTTTCCTTGGGGT-3′ |
| NF-κB | 5′-GTATTTCAACCACAGATGGCACT-3′ | 5′-AACCTTTGCTGGTCCCACAT-3′ |
| RANTES | 5′-GCGGGTACCATGAAGATCTCTG-3′ | 5′-GGGTCAGAATCAAGAAACCCTC-3′ |
| IFN-α | 5′-TTTCTCCTGCCTGAAGGACAG-3′ | 5′-GCTCATGATTTCTGCTCTGACA-3′ |
| IL-1β | 5′-GTGCAGTTTTGCCAAGGAG-3′ | 5′-ACGTTTCGAAGATGACAGGCT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beirag, N.; Varghese, P.M.; Neto, M.M.; Al Aiyan, A.; Khan, H.A.; Qablan, M.; Shamji, M.H.; Sim, R.B.; Temperton, N.; Kishore, U. Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein. Viruses 2023, 15, 1269. https://doi.org/10.3390/v15061269
Beirag N, Varghese PM, Neto MM, Al Aiyan A, Khan HA, Qablan M, Shamji MH, Sim RB, Temperton N, Kishore U. Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein. Viruses. 2023; 15(6):1269. https://doi.org/10.3390/v15061269
Chicago/Turabian StyleBeirag, Nazar, Praveen M. Varghese, Martin Mayora Neto, Ahmad Al Aiyan, Haseeb A. Khan, Moneeb Qablan, Mohamed H. Shamji, Robert B. Sim, Nigel Temperton, and Uday Kishore. 2023. "Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein" Viruses 15, no. 6: 1269. https://doi.org/10.3390/v15061269
APA StyleBeirag, N., Varghese, P. M., Neto, M. M., Al Aiyan, A., Khan, H. A., Qablan, M., Shamji, M. H., Sim, R. B., Temperton, N., & Kishore, U. (2023). Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein. Viruses, 15(6), 1269. https://doi.org/10.3390/v15061269