

Recent Population Dynamics of Japanese Encephalitis Virus
 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
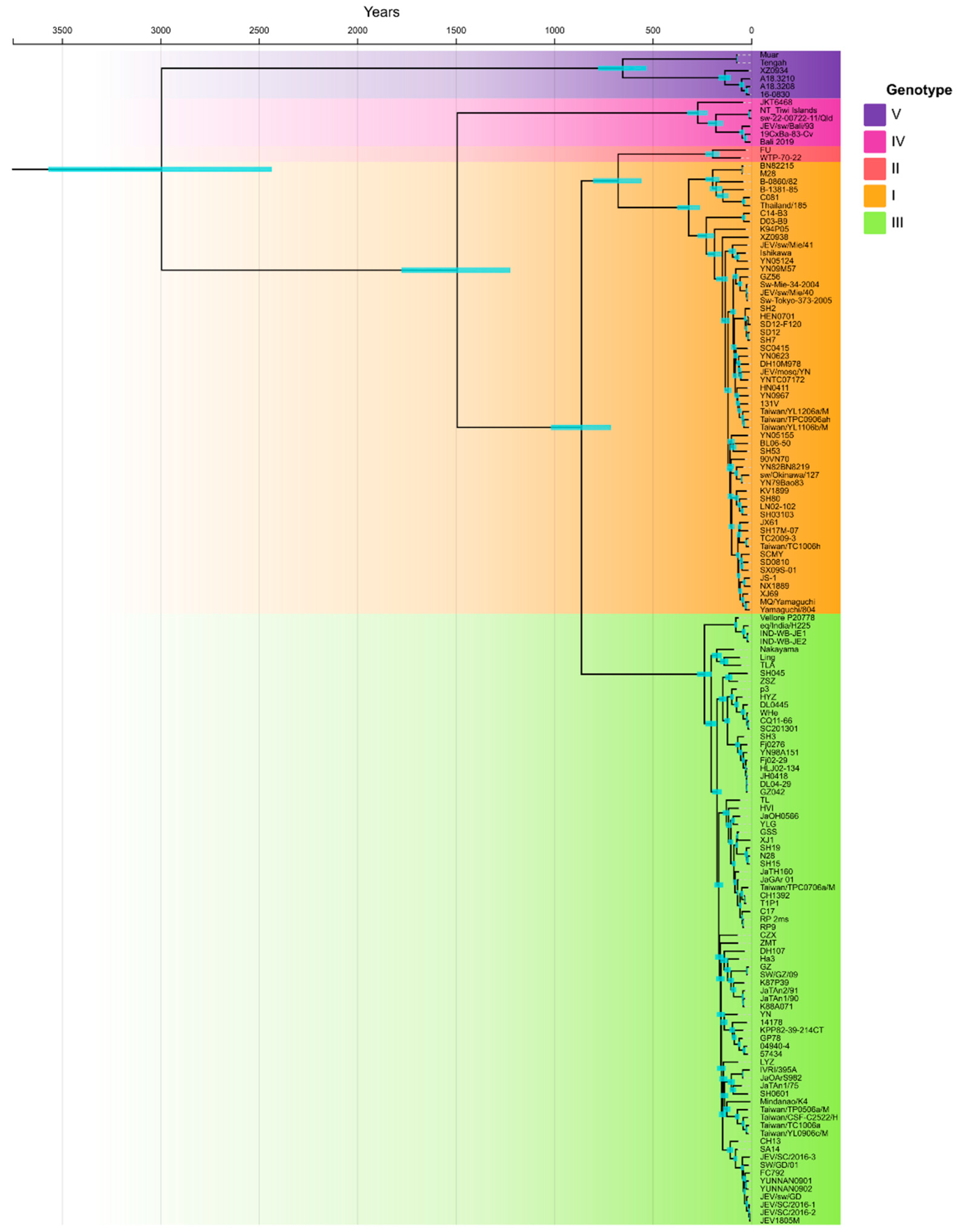
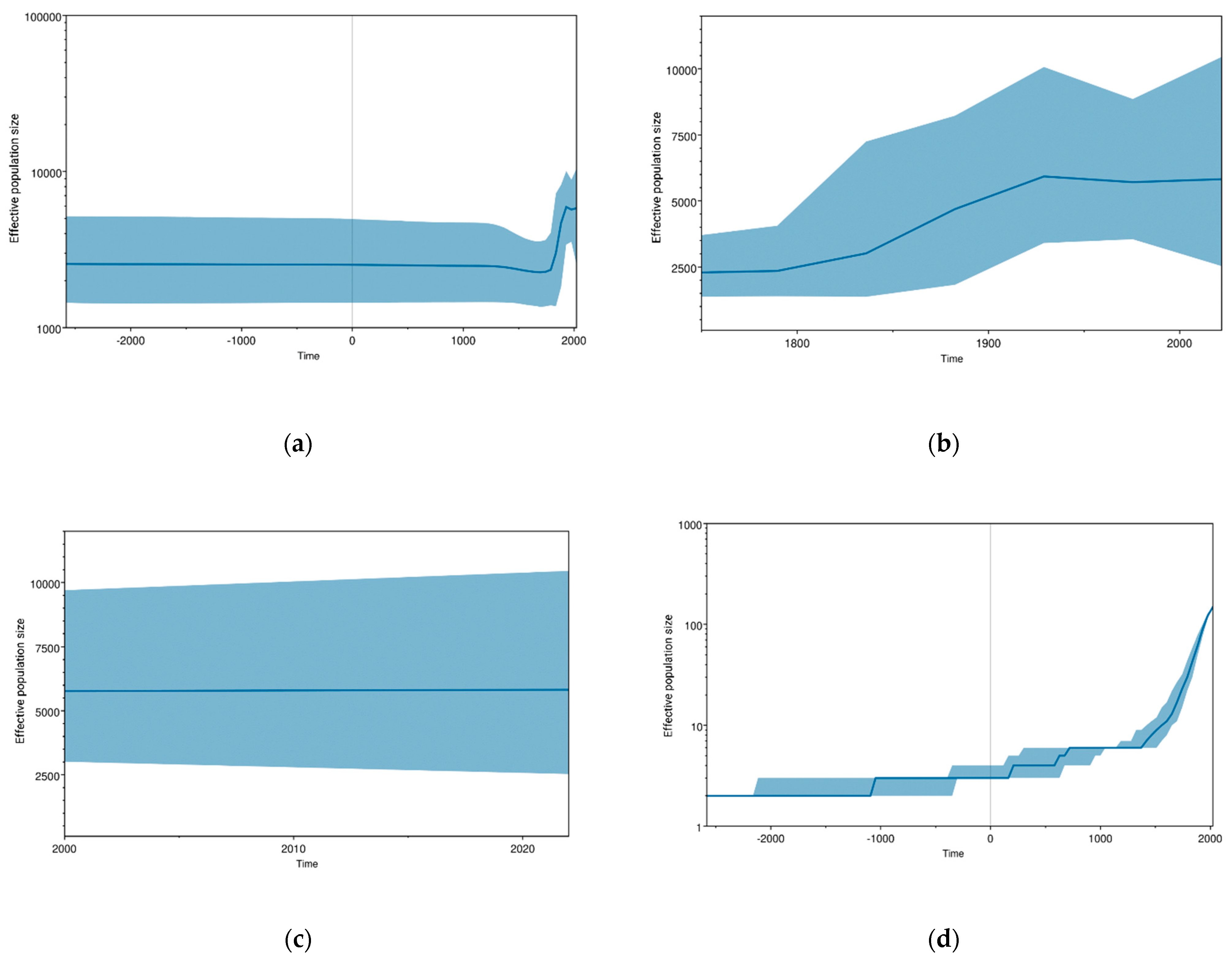
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campbell, G.L.; Hills, S.L.; Fischer, M.; Jacobson, J.A.; Hoke, C.H.; Hombach, J.M.; Marfin, A.A.; Solomon, T.; Tsai, T.F.; Tsu, V.D.; et al. Estimated global incidence of Japanese encephalitis: A systematic review. Bull. World Health Organ. 2011, 89, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.J.; Ward, M.J.; Brown, A.L.; Barrett, A.D.T. Phylogeography of Japanese encephalitis virus: Genotype is associated with climate. PLoS Negl. Trop. Dis. 2013, 7, e2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, M.; Liu, K.; Anwar, M.N.; Wahaab, A.; Safdar, A.; Di, D.; Boruah, P.; Xu, J.; Wang, X.; Li, B.; et al. The emerged genotype I of Japanese encephalitis virus shows an infectivity similar to genotype III in Culex pipiens mosquitoes from China. PLoS Negl. Trop. Dis. 2019, 13, e0007716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, M.; Wahaab, A.; Nawaz, M.; Khan, S.; Nazir, J.; Liu, K.; Wei, J.; Ma, Z. Potential Role of Birds in Japanese Encephalitis Virus Zoonotic Transmission and Genotype Shift. Viruses 2021, 13, 357. [Google Scholar] [CrossRef]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Misra, U.K.; Kalita, J. Overview: Japanese encephalitis. Prog. Neurobiol. 2010, 91, 108–120. [Google Scholar] [CrossRef]
- Heffelfinger, J.D.; Li, X.; Batmunkh, N.; Grabovac, V.; Diorditsa, S.; Liyanage, J.B.; Pattamadilok, S.; Bahl, S.; Vannice, K.S.; Hyde, T.B.; et al. Japanese Encephalitis Surveillance and Immunization—Asia and Western Pacific Regions, 2016. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 579–583. [Google Scholar] [CrossRef]
- Gao, X.; Liu, H.; Li, M.; Fu, S.; Liang, G. Insights into the evolutionary history of Japanese encephalitis virus (JEV) based on whole-genome sequences comprising the five genotypes. Virol. J. 2015, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Gao, T.; Wang, Z.; Zhang, J.; Cui, B.; Shen, X.; Zhou, A.; Zhang, Y.; Zhao, J.; Liu, H.; et al. Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution. Viruses 2023, 15, 626. [Google Scholar] [CrossRef]
- Zhang, H.; Rehman, M.U.; Li, K.; Luo, H.; Lan, Y.; Nabi, F.; Zhang, L.; Iqbal, M.K.; Zhu, S.; Javed, M.T.; et al. Epidemiologic Survey of Japanese Encephalitis Virus Infection, Tibet, China, 2015. Emerg. Infect. Dis. 2017, 23, 1023–1024. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, H.; Rehman, M.U.; Nabi, F.; Li, K.; Lan, Y.; Huang, S.; Zhang, L.; Mehmood, K.; Shahzad, M.; et al. Evidence of JEV in Culex tritaeniorhynchus and pigs from high altitude regions of Tibet, China. J. Vector Borne Dis. 2017, 54, 69–73. [Google Scholar] [PubMed]
- Aure, W.E.; Sayama, Y.; Saito-Obata, M.; Salazar, N.P.; Malbas, F.F.; Galang, H.O.; Imamura, T.; Zuasula, C.L.; Oshitani, H. Japanese encephalitis virus genotype III from mosquitoes in Tarlac, Philippines. IJID Reg. 2022, 4, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Howard-Jones, A.R.; Pham, D.; Jeoffreys, N.; Eden, J.-S.; Hueston, L.; Kesson, A.M.; Nagendra, V.; Samarasekara, H.; Newton, P.; Chen, S.C.-A.; et al. Emerging Genotype IV Japanese Encephalitis Virus Outbreak in New South Wales, Australia. Viruses 2022, 14, 1853. [Google Scholar] [CrossRef] [PubMed]
- Waller, C.; Tiemensma, M.; Currie, B.J.; Williams, D.T.; Baird, R.W.; Krause, V.L. Japanese Encephalitis in Australia—A Sentinel Case. N. Engl. J. Med. 2022, 387, 661–662. [Google Scholar] [CrossRef]
- Liu, W.; Fu, S.; Ma, X.; Chen, X.; Wu, D.; Zhou, L.; Yin, Q.; Li, F.; He, Y.; Lei, W.; et al. An outbreak of Japanese encephalitis caused by genotype Ib Japanese encephalitis virus in China, 2018: A laboratory and field investigation. PLoS Negl. Trop. Dis. 2020, 14, e0008312. [Google Scholar] [CrossRef]
- Fang, Y.; Li, X.-S.; Zhang, W.; Xue, J.-B.; Wang, J.-Z.; Yin, S.-Q.; Li, S.-G.; Li, X.-H.; Zhang, Y. Molecular epidemiology of mosquito-borne viruses at the China–Myanmar border: Discovery of a potential epidemic focus of Japanese encephalitis. Infect. Dis. Poverty 2021, 10, 57. [Google Scholar] [CrossRef]
- Hameed, M.; Wahaab, A.; Shan, T.; Wang, X.; Khan, S.; Di, D.; Xiqian, L.; Zhang, J.-J.; Anwar, M.N.; Nawaz, M.; et al. A Metagenomic Analysis of Mosquito Virome Collected From Different Animal Farms at Yunnan–Myanmar Border of China. Front. Microbiol. 2020, 11, 591478. [Google Scholar] [CrossRef]
- Hameed, M.; Khan, S.; Xu, J.; Zhang, J.; Wang, X.; Di, D.; Chen, Z.; Anwar, M.N.; Wahaab, A.; Ma, X.; et al. Detection of Japanese encephalitis virus in mosquitoes from Xinjiang during next-generation sequencing arboviral surveillance. Transbound. Emerg. Dis. 2021, 68, 467–476. [Google Scholar] [CrossRef]
- Sanborn, M.A.; Wuertz, K.M.; Heung-Chul, K.; Yang, Y.; Li, T.; Pollett, S.D.; Jarman, R.G.; Berry, I.M.; Klein, T.A.; Hang, J. Identification of Japanese Encephalitis Virus Genotype V and Other Mosquito-borne Viruses in Camp Humphreys, Republic of Korea, using Metagenomic Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mackenzie, J.S.; Williams, D.T.; Hurk, A.F.v.D.; Smith, D.W.; Currie, B.J. Japanese Encephalitis Virus: The Emergence of Genotype IV in Australia and Its Potential Endemicity. Viruses 2022, 14, 2480. [Google Scholar] [CrossRef]
- Hurk, A.F.V.D.; Skinner, E.; Ritchie, S.A.; Mackenzie, J.S. The Emergence of Japanese Encephalitis Virus in Australia in 2022: Existing Knowledge of Mosquito Vectors. Viruses 2022, 14, 1208. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. (Ed.) BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT; Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; pp. c1979–c2000. [Google Scholar]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.; Solomon, W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, L.; Taylor, H.G. Japanese B encephalitis; clinical observations in an outbreak on Okinawa Shima. Arch. Neurol. Psychiatry 1947, 57, 430–463. [Google Scholar] [CrossRef]
- Gao, X.; Nasci, R.; Liang, G. The neglected arboviral infections in mainland China. PLoS Negl. Trop. Dis. 2010, 4, e624. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y. Phenotypic and genotypic characteristics of Japanese encephalitis attenuated live vaccine virus SA14-14-2 and their stabilities. Vaccine 2010, 28, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-L.; Liu, H.; Wang, H.-Y.; Fu, S.-H.; Liu, H.-Z.; Zhang, H.-L.; Li, M.-H.; Gao, X.-Y.; Wang, J.-L.; Sun, X.-H.; et al. Emergence of genotype I of Japanese encephalitis virus as the dominant genotype in Asia. J. Virol. 2011, 85, 9847–9853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-J.S.; Higgs, S.; Vanlandingham, D.L. Emergence and re-emergence of mosquito-borne arboviruses. Curr. Opin. Virol. 2019, 34, 104–109. [Google Scholar] [CrossRef]
- Peinado, R.D.S.; Eberle, R.J.; Arni, R.K.; Coronado, M.A. A Review of Omics Studies on Arboviruses: Alphavirus, Orthobunyavirus and Phlebovirus. Viruses 2022, 14, 2194. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, S.; Di Giallonardo, F.; Holmes, E.C. Substitution Model Adequacy and Assessing the Reliability of Estimates of Virus Evolutionary Rates and Time Scales. Mol. Biol. Evol. 2016, 33, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Añez, G.; Grinev, A.; Chancey, C.; Ball, C.; Akolkar, N.; Land, K.J.; Winkelman, V.; Stramer, S.L.; Kramer, L.D.; Rios, M. Evolutionary dynamics of West Nile virus in the United States, 1999–2011: Phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 2013, 7, e2245. [Google Scholar] [CrossRef] [Green Version]
- Egyed, L.; Rónai, Z.; Dán, Á. Hungarian tickborne encephalitis viruses isolated from a 0.5-ha focus are closely related to Finnish strains. Ticks Tick-Borne Dis. 2018, 9, 1064–1068. [Google Scholar] [CrossRef]
- Bryant, J.E.; Holmes, E.C.; Barrett, A.D.T. Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007, 3, e75. [Google Scholar] [CrossRef] [Green Version]
- Garjito, T.A.; Widiarti; Anggraeni, Y.M.; Alfiah, S.; Satoto, T.B.T.; Farchanny, A.; Samaan, G.; Afelt, A.; Manguin, S.; Frutos, R.; et al. Japanese encephalitis in Indonesia: An update on epidemiology and transmission ecology. Acta Trop. 2018, 187, 240–247. [Google Scholar] [CrossRef]
- Solomon, T.; Ni, H.; Beasley, D.W.C.; Ekkelenkamp, M.; Cardosa, M.J.; Barrett, A.D.T. Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 2003, 77, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Li, C.; Di, D.; Cappelle, J.; Liu, L.; Wang, X.; Pang, L.; Xu, J.; Liu, K.; Li, B.; et al. Differential replication efficiencies between Japanese encephalitis virus genotype I and III in avian cultured cells and young domestic ducklings. PLoS Negl. Trop. Dis. 2018, 12, e0007046. [Google Scholar] [CrossRef] [Green Version]
- Palm, E.C.; Newman, S.H.; Prosser, D.J.; Xiao, X.; Ze, L.; Batbayar, N.; Balachandran, S.; Takekawa, J.Y. Mapping migratory flyways in Asia using dynamic Brownian bridge movement models. Mov. Ecol. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Bae, W.; Kim, J.H.; Kim, J.; Lee, J.; Hwang, E.S. Changes of epidemiological characteristics of Japanese encephalitis viral infection and birds as a potential viral transmitter in Korea. J. Korean Med. Sci. 2018, 33, e70. [Google Scholar] [CrossRef] [PubMed]
- Marini, G.; Manica, M.; Arnoldi, D.; Inama, E.; Rosà, R.; Rizzoli, A. Influence of Temperature on the Life-Cycle Dynamics of Aedes albopictus Population Established at Temperate Latitudes: A Laboratory Experiment. Insects 2020, 11, 808. [Google Scholar] [CrossRef] [PubMed]
- Roche, B.; Léger, L.; L’ambert, G.; Lacour, G.; Foussadier, R.; Besnard, G.; Barré-Cardi, H.; Simard, F.; Fontenille, D. The Spread of Aedes albopictus in Metropolitan France: Contribution of Environmental Drivers and Human Activities and Predictions for a Near Future. PLoS ONE 2015, 10, e0125600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuno, N.; Tsuda, Y.; Takagi, M. How Zoophilic Japanese Encephalitis Vector Mosquitoes Feed on Humans. J. Med. Èntomol. 2017, 54, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hu, W.; Magalhaes, R.J.S.; Bi, P.; Ding, F.; Sun, H.; Li, S.; Yin, W.; Wei, L.; Liu, Q.; et al. The role of environmental factors in the spatial distribution of Japanese encephalitis in mainland China. Environ. Int. 2014, 73, 1–9. [Google Scholar] [CrossRef]
- Tu, T.; Xu, K.; Xu, L.; Gao, Y.; Zhou, Y.; He, Y.; Liu, Y.; Liu, Q.; Ji, H.; Tang, W. Association between meteorological factors and the prevalence dynamics of Japanese encephalitis. PLoS ONE 2021, 16, e0247980. [Google Scholar] [CrossRef]
- Srivastava, K.S.; Jeswani, V.; Pal, N.; Bohra, B.; Vishwakarma, V.; Bapat, A.A.; Patnaik, Y.P.; Khanna, N.; Shukla, R. Japanese Encephalitis Virus: An Update on the Potential Antivirals and Vaccines. Vaccines 2023, 11, 742. [Google Scholar] [CrossRef]
- Mileno, M.D. Japanese Encephalitis Vaccine. Rhode Isl. Med. J. 2020, 103, 49–50. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Wahaab, A.; Khan, S.; Nawaz, M.; Anwar, M.N.; Liu, K.; Wei, J.; Hameed, M.; Ma, Z. Recent Population Dynamics of Japanese Encephalitis Virus. Viruses 2023, 15, 1312. https://doi.org/10.3390/v15061312
Xu J, Wahaab A, Khan S, Nawaz M, Anwar MN, Liu K, Wei J, Hameed M, Ma Z. Recent Population Dynamics of Japanese Encephalitis Virus. Viruses. 2023; 15(6):1312. https://doi.org/10.3390/v15061312
Chicago/Turabian StyleXu, Jinpeng, Abdul Wahaab, Sawar Khan, Mohsin Nawaz, Muhammad Naveed Anwar, Ke Liu, Jianchao Wei, Muddassar Hameed, and Zhiyong Ma. 2023. "Recent Population Dynamics of Japanese Encephalitis Virus" Viruses 15, no. 6: 1312. https://doi.org/10.3390/v15061312
APA StyleXu, J., Wahaab, A., Khan, S., Nawaz, M., Anwar, M. N., Liu, K., Wei, J., Hameed, M., & Ma, Z. (2023). Recent Population Dynamics of Japanese Encephalitis Virus. Viruses, 15(6), 1312. https://doi.org/10.3390/v15061312