
Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020

 , , , , , ,
, , , , , , 
Abstract
:1. Introduction
Objectives
2. Materials and Methods
2.1. Cohort Study and Clinical Samples
2.2. Detection of Enterovirus Infections Based on Real-Time RT PCR
2.3. Genotyping Enterovirus A and B Based on 5′-UTR Sequence Analysis
2.4. Viral Whole Genome Sequencing for a Double Infection Case
2.5. Nanopore Sequencing Data Analysis
3. Results
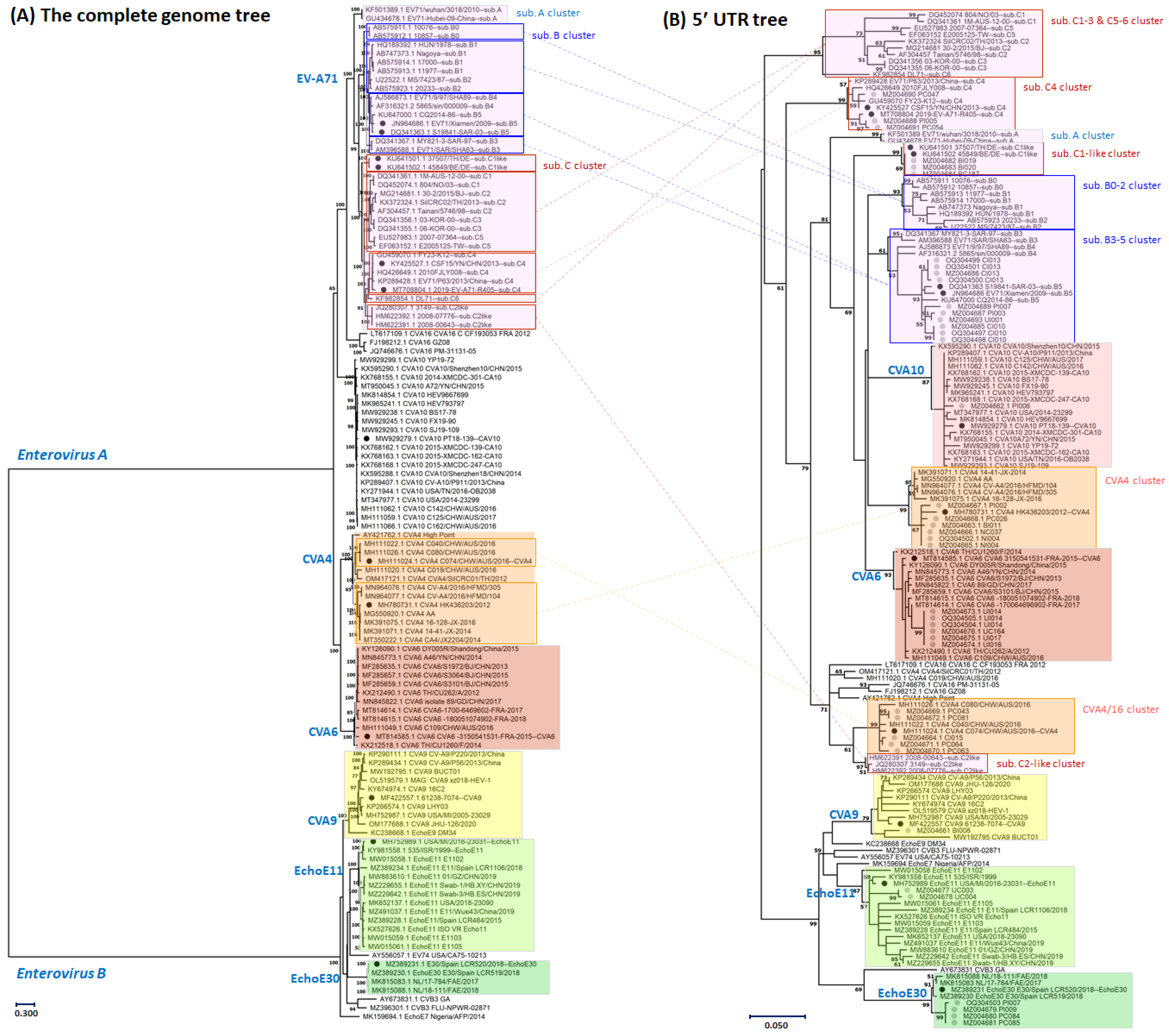
3.1. Genotyping of the Enterovirus Infections Based on Partial Sequences of the Five Prime Untranslated Region
3.2. Statistics of Enterovirus Infections and Manifestations in the Cohort Study
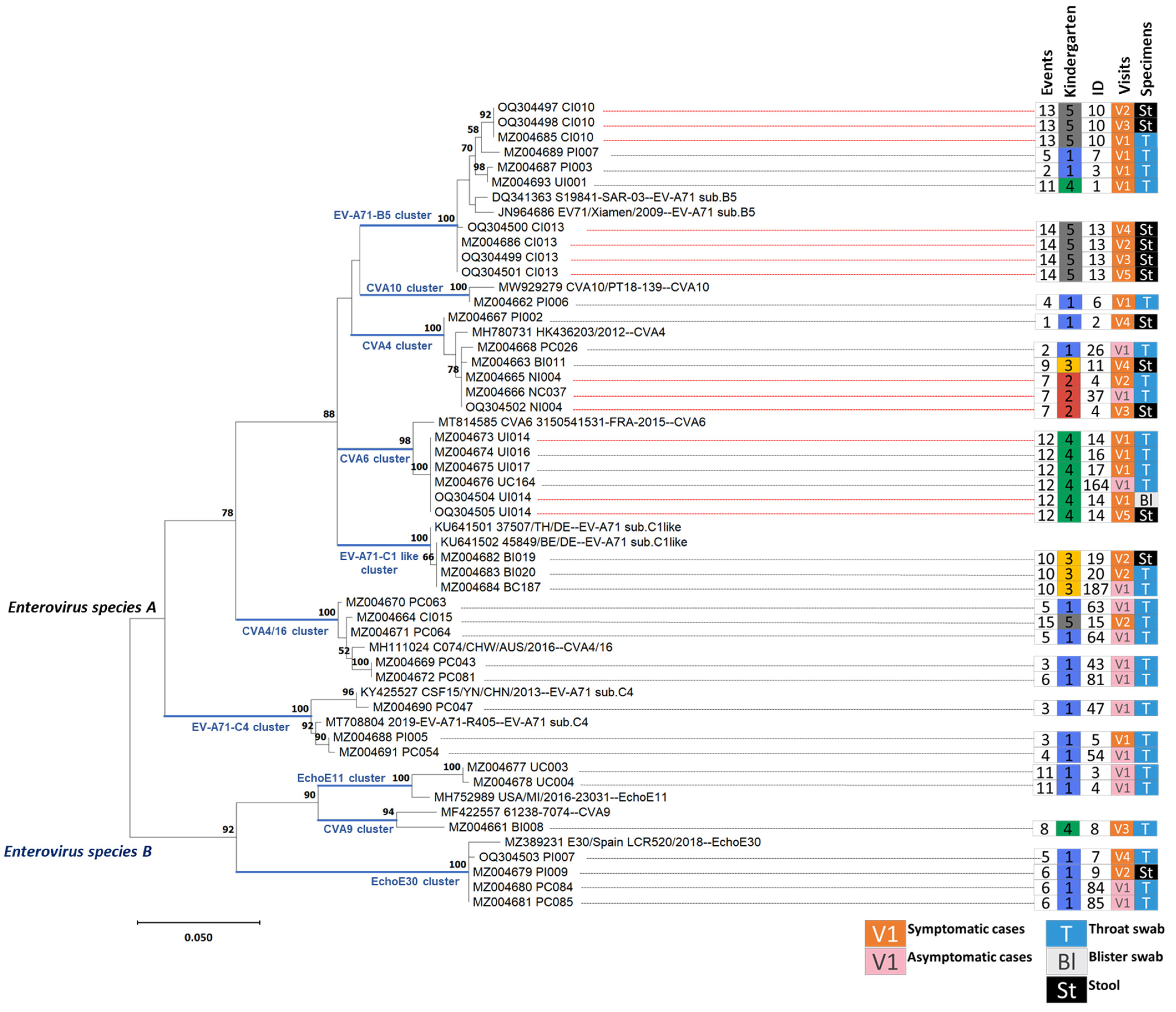
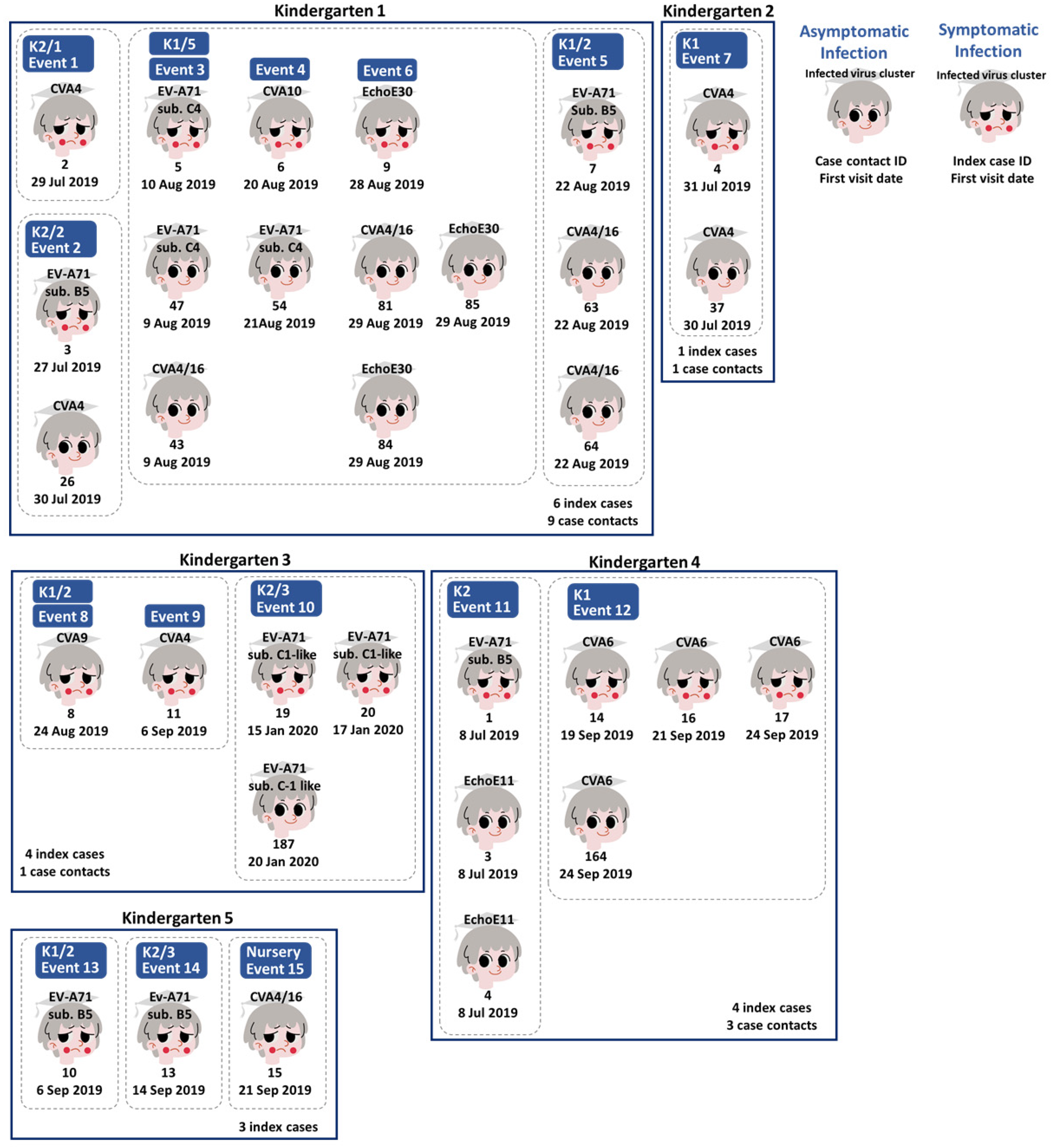
3.3. Human Enterovirus Infections Circulating in Five Kindergartens
3.4. Enteroviral RNA Shedding in Throat Swabs and Stool Samples of Index Cases
3.5. Viral Whole Genome Sequencing for Tracking Echovirus E30 Transmission between Classrooms in the Same Kindergarten
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corsino, C.B.; Ali, R.; Linklater, D.R. Herpangina. In StatPearls; StatPearls Publishing Copyright © 2023; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Li, W.; Gao, H.-H.; Zhang, Q.; Liu, Y.-J.; Tao, R.; Cheng, Y.-P.; Shu, Q.; Shang, S.-Q. Large outbreak of herpangina in children caused by enterovirus in summer of 2015 in Hangzhou, China. Sci. Rep. 2016, 6, 35388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ceano-Vivas, M.; Garcia, M.L.; Velazquez, A.; Martin Del Valle, F.; Menasalvas, A.; Cilla, A.; Epalza, C.; Romero, M.P.; Cabrerizo, M.; Calvo, C. Neurodevelopmental Outcomes of Infants Younger Than 90 Days Old Following Enterovirus and Parechovirus Infections of the Central Nervous System. Front. Pediatr. 2021, 9, 719119. [Google Scholar] [CrossRef] [PubMed]
- Olchawa-Czech, A.; Ptak, K.; Szymonska, I.; Kwinta, P. Severe enterovirus infections in infants <3 months of age and the importance of medical history. J. Mother. Child. 2021, 24, 37–44. [Google Scholar] [PubMed]
- Yang, X.; Duan, L.; Zhan, W.; Tang, Y.; Liang, L.; Xie, J.; Luo, M. Enterovirus B types cause severe infection in infants aged 0–3 months. Virol. J. 2023, 20, 5. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Chen, W.; He, W.; Huang, M.; Zhu, Y.; Yan, Y. Serotyping and Genetic Characterization of Hand, Foot, and Mouth Disease (HFMD)-Associated Enteroviruses of No-EV71 and Non-CVA16 Circulating in Fujian, China, 2011–2015. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Nhan, L.N.T.; Turner, H.C.; Khanh, T.H.; Hung, N.T.; Lien, L.B.; Hong, N.T.T.; Nhu, L.N.T.; Ny, N.T.H.; Nguyet, L.A.; Thanh, T.T.; et al. Economic Burden Attributed to Children Presenting to Hospitals with Hand, Foot, and Mouth Disease in Vietnam. Open Forum Infect. Dis. 2019, 6, ofz284. [Google Scholar] [CrossRef]
- Lerdsamran, H.; Prasertsopon, J.; Mungaomklang, A.; Klinmalai, C.; Noisumdaeng, P.; Sangsiriwut, K.; Tassaneetrithep, B.; Guntapong, R.; Iamsirithaworn, S.; Puthavathana, P. Seroprevalence of antibodies to enterovirus 71 and coxsackievirus A16 among people of various age groups in a northeast province of Thailand. Virol. J. 2018, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Linsuwanon, P.; Puenpa, J.; Huang, S.-W.; Wang, Y.-F.; Mauleekoonphairoj, J.; Wang, J.-R.; Poovorawan, Y. Epidemiology and seroepidemiology of human enterovirus 71 among Thai populations. J. Biomed. Sci. 2014, 21, 16. [Google Scholar] [CrossRef] [Green Version]
- Puenpa, J.; Auphimai, C.; Korkong, S.; Vongpunsawad, S.; Poovorawan, Y. Enterovirus A71 Infection, Thailand, 2017. Emerg. Infect. Dis. 2018, 24, 1386–1387. [Google Scholar] [CrossRef] [Green Version]
- Puenpa, J.; Chieochansin, T.; Linsuwanon, P.; Korkong, S.; Thongkomplew, S.; Vichaiwattana, P.; Theamboonlers, A.; Poovorawan, Y. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg. Infect. Dis. 2013, 19, 641–643. [Google Scholar] [CrossRef] [Green Version]
- Organisation, W.H. Enterovirus Infection—France. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON469 (accessed on 14 June 2023).
- Chen, M.; Zuo, X.; Tan, Y.; Ju, Y.; Bi, F.; Wang, H.; Chen, M. Six amino acids of VP1 switch along with pandemic of CV-A6-associated HFMD in Guangxi, southern China, 2010–2017. J. Infect. 2019, 78, 323–337. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X.; et al. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci. Rep. 2017, 7, 5491. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, Y.; Zhang, C.; Zhan, W.; Xie, J.; Hu, S.; Chai, H.; Liu, P.; Zhao, H.; Tang, B.; et al. Clinical features and phylogenetic analysis of severe hand-foot-and-mouth disease caused by Coxsackievirus A6. Infect. Genet. Evol. 2020, 77, 104054. [Google Scholar] [CrossRef]
- Gauthier, N.P.G.; Nelson, C.; Bonsall, M.B.; Locher, K.; Charles, M.; MacDonald, C.; Krajden, M.; Chorlton, S.D.; Manges, A.R. Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples. PLoS ONE 2021, 16, e0259712. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef] [Green Version]
- Wollants, E.; Maes, P.; Merino, M.; Bloemen, M.; Van Ranst, M.; Vanmechelen, B. First genomic characterization of a Belgian Enterovirus C104 using sequence-independent Nanopore sequencing. Infect. Genet. Evol. 2020, 81, 104267. [Google Scholar] [CrossRef]
- Zakotnik, S.; Korva, M.; Knap, N.; Robnik, B.; Gorišek Miksić, N.; Avšič Županc, T. Complete Coding Sequence of a Chikungunya Virus Strain Imported into Slovenia from Thailand in Late 2018. Microbiol. Resour. Announc. 2019, 8, e00581-19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhao, L.; Zhang, Z.; Hong, W.; Wang, J.; Qiu, S.; Yang, H.; Gan, M.; Sun, J.; Zhao, J.; et al. Genetic and pathogenicity diversity of dengue virus type 2 strains circulating in Guangdong, China. Biosaf. Health 2021, 3, 333–342. [Google Scholar] [CrossRef]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef] [PubMed]
- Thammasonthijarern, N.; Kosoltanapiwat, N.; Nuprasert, W.; Sittikul, P.; Sriburin, P.; Pan-Ngum, W.; Maneekan, P.; Hataiyusuk, S.; Hattasingh, W.; Thaipadungpanit, J.; et al. Molecular Epidemiological Study of Hand, Foot, and Mouth Disease in a Kindergarten-Based Setting in Bangkok, Thailand. Pathogens 2021, 10, 576. [Google Scholar] [CrossRef] [PubMed]
- Lekana-Douki, S.E.; Sir-Ondo-Enguier, P.N.; Banga-Mve-Ella, O.; Imboumy-Limoukou, R.K.; Maganga, G.D.; Lekana-Douki, J.B.; Berthet, N. Epidemiology and molecular characterization of the re-emerging measles virus among children and adults in the Haut-Ogooue, Gabon. BMC Infect. Dis. 2019, 19, 90. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grüning, B.; Dale, R.; Sjödin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Köster, J. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.Y.; Liu, J.; Lai, W.; Luo, J.; Liu, Y.; Vu, G.P.; Yang, Z.; Trang, P.; Li, H.; Wu, J. Characterization of enterovirus 71 infection and associated outbreak of Hand, Foot, and Mouth Disease in Shawo of China in 2012. Sci. Rep. 2016, 6, 38451. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.J.; Romero, J.R.; Wasserman, R.; Rotbart, H.A. Stable Enterovirus 5′ Nontranslated Region over a 7-Year Period in a Patient with Agammaglobulinemia and Chronic Infection. J. Infect. Dis. 2000, 182, 298–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; He, S.; Yan, Q.; Xu, X.; Wu, W.; Ge, S.; Zhang, S.; Chen, M.; Xia, N. Severe hand, foot and mouth disease associated with Coxsackievirus A10 infections in Xiamen, China in 2015. J. Clin. Virol. 2017, 93, 20–24. [Google Scholar] [CrossRef]
- Yang, F.; Yuan, J.; Wang, X.; Li, J.; Du, J.; Su, H.; Zhou, B.; Jin, Q. Severe Hand, Foot, and Mouth Disease and Coxsackievirus A6—Shenzhen, China. Clin. Infect. Dis. 2014, 59, 1504–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noisumdaeng, P.; Korkusol, A.; Prasertsopon, J.; Sangsiriwut, K.; Chokephaibulkit, K.; Mungaomklang, A.; Thitithanyanont, A.; Buathong, R.; Guntapong, R.; Puthavathana, P. Longitudinal study on enterovirus A71 and coxsackievirus A16 genotype/subgenotype replacements in hand, foot and mouth disease patients in Thailand, 2000–2017. Int. J. Infect. Dis. 2019, 80, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.T.; Kobayashi, K.; Bi, X.; Ishizaki, A.; Tran, T.T.; Phung, T.T.B.; Pham, C.T.T.; Nguyen, L.V.; Ta, T.A.; Khu, D.T.K.; et al. Newly emerged enterovirus-A71 C4 sublineage may be more virulent than B5 in the 2015–2016 hand-foot-and-mouth disease outbreak in northern Vietnam. Sci. Rep. 2020, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, J.; Ji, G.; Gao, Y.; Zhang, C.; Zhang, T.; Huo, J.; Liang, W.; Yang, J.; Shi, Y.; et al. A novel subgenotype C6 Enterovirus A71 originating from the recombination between subgenotypes C4 and C2 strains in mainland China. Sci. Rep. 2022, 12, 593. [Google Scholar] [CrossRef]
- Zeng, H.; Yi, L.; Chen, X.; Zhou, H.; Zheng, H.; Lu, J.; Yang, F.; Li, C.; Fang, L.; Zhang, X.; et al. Emergence of a non vaccine-cognate enterovirus A71 genotype C1 in mainland China. J. Infect. 2021, 82, 407–413. [Google Scholar] [CrossRef]
- Li, J.; Ni, N.; Cui, Y.; Zong, S.; Yao, X.; Hu, T.; Cao, M.; Zhang, Y.; Hou, P.; Carr, M.J.; et al. An outbreak of a novel recombinant Coxsackievirus A4 in a kindergarten, Shandong province, China, 2021. Emerg. Microbes Infect. 2022, 11, 2207–2210. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Yao, M.-X.; Zhang, Y.-W.; Hu, T.; Carr, M.J.; Duchêne, S.; Zhang, X.-C.; Zhang, Z.-J.; Zhou, H.; et al. Genome Analysis of Coxsackievirus A4 Isolates from Hand, Foot, and Mouth Disease Cases in Shandong, China. Front. Microbiol. 2019, 10, 1001. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, L.; Moreni, G.; Wolthers, K.C.; Pajkrt, D. World-Wide Prevalence and Genotype Distribution of Enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef]
- CDC. Outbreaks of Aseptic Meningitis Associated with Echoviruses 9 and 30 and Preliminary Surveillance Reports on Enterovirus Activity—United States, 2003. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5232a1.htm (accessed on 16 May 2023).
- Li, J.; Yan, D.; Chen, L.; Zhang, Y.; Song, Y.; Zhu, S.; Ji, T.; Zhou, W.; Gan, F.; Wang, X.; et al. Multiple genotypes of Echovirus 11 circulated in mainland China between 1994 and 2017. Sci. Rep. 2019, 9, 10583. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Han, Z.; He, Y.; Sun, Q.; Wang, W.; Xu, W.; Li, H.; Zhang, Y. Temporal phylogeny and molecular characterization of echovirus 30 associated with aseptic meningitis outbreaks in China. Virol. J. 2021, 18, 118. [Google Scholar] [CrossRef]
- Xiao, H.; Guan, D.; Chen, R.; Chen, P.; Monagin, C.; Li, W.; Su, J.; Ma, C.; Zhang, W.; Ke, C. Molecular characterization of echovirus 30-associated outbreak of aseptic meningitis in Guangdong in 2012. Virol. J. 2013, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyoshi, K.; Nakagawa, N.; Suga, T. An outbreak of aseptic meningitis in a nursery school caused by echovirus type 30 in Kobe, Japan. Jpn. J. Infect. Dis. 2007, 60, 66–68. [Google Scholar]
- Zheng, S.; Ye, H.; Yan, J.; Xie, G.; Cui, D.; Yu, F.; Wang, Y.; Yang, X.; Zhou, F.; Zhang, Y.; et al. Laboratory diagnosis and genetic analysis of a family clustering outbreak of aseptic meningitis due to echovirus 30. Pathog. Glob. Health 2016, 110, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Hemachudha, P.; Petcharat, S.; Hinjoy, S.; Saraya, A.W.; Hemachudha, T. Encephalitis in Thailand: A Neglected Disease Increasingly Caused by Enterovirus. Trop. Med. Infect. Dis. 2021, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Nagington, J.; Wreghittt, T.G.; Gandy, G.; Roberton, N.R.; Berry, P.J. Fatal echovirus 11 infections in outbreak in special-care baby unit. Lancet 1978, 312 Pt 1, 725–728. [Google Scholar] [CrossRef]
- Apisarnthanarak, A.; Kitphati, R.; Pongsuwann, Y.; Tacharoenmueng, R.; Mundy, L.M. Echovirus type 11: Outbreak of hand-foot-and-mouth disease in a Thai hospital nursery. Clin. Infect. Dis. 2005, 41, 1361–1362. [Google Scholar] [CrossRef]
- Lim, C.T.; Jiang, L.; Ma, S.; James, L.; Ang, L.W. Basic reproduction number of coxsackievirus type A6 and A16 and enterovirus 71: Estimates from outbreaks of hand, foot and mouth disease in Singapore, a tropical city-state. Epidemiol. Infect. 2016, 144, 1028–1034. [Google Scholar] [CrossRef]
- Marotta, C.; Di Gennaro, F.; Pizzol, D.; Madeira, G.; Monno, L.; Saracino, A.; Putoto, G.; Casuccio, A.; Mazzucco, W. The At Risk Child Clinic (ARCC): 3 Years of Health Activities in Support of the Most Vulnerable Children in Beira, Mozambique. Int. J. Environ. Res. Public Health 2018, 15, 1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Ma, X.-J.; Wan, J.-F.; Liu, Y.-H.; Han, Y.-L.; Chen, C.; Tian, C.; Gao, C.; Wang, M.; Dong, X.-P. Long persistence of EV71 specific nucleotides in respiratory and feces samples of the patients with Hand-Foot-Mouth Disease after recovery. BMC Infect. Dis. 2010, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lin, C.; Qu, M.; Li, X.; Gao, Z.; Zhang, X.; Liu, Y.; Huang, Y.; Wang, X.; Jia, L.; et al. Excretion of enterovirus 71 in persons infected with hand, foot and mouth disease. Virol. J. 2013, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Liao, Q.; Ooi, M.H.; Cowling, B.J.; Chang, Z.; Wu, P.; Liu, F.; Li, Y.; Luo, L.; Yu, S.; et al. Epidemiology of Recurrent Hand, Foot and Mouth Disease, China, 2008–2015. Emerg. Infect. Dis. 2018, 24, 432–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Liu, J.; Shi, P.; Ji, H.; Shen, Y.; Qian, Y.H. Epidemiological characteristics and influential factors of hand, foot, and mouth disease reinfection in Wuxi, China, 2008–2016. BMC Infect. Dis. 2018, 18, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Wang, H.; Chen, C.; Zou, X.; Li, T. Epidemiological Characteristics of Hand, Foot and Mouth Disease Reinfection in Guangzhou, Southern China from 2012 to 2017. Iran J. Public Health 2022, 51, 2078–2088. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, J.; Chen, J.; Huang, R.; Zhang, Y.; Xiao, J.; Song, Y.; Ji, T.; Yang, Q.; Zhu, S.; et al. Molecular Epidemiology and Evolution of Coxsackievirus A9. Viruses 2022, 14, 822. [Google Scholar] [CrossRef]
- Liu, M.Y.; Liu, W.; Luo, J.; Liu, Y.; Zhu, Y.; Berman, H.; Wu, J. Characterization of an outbreak of hand, foot, and mouth disease in Nanchang, China in 2010. PLoS ONE 2011, 6, e25287. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Enterovirus Clusters | a Infection Distributions (K. 1-2-3-4-5) | b Age (Min–Max) | c Participants with ASI (Ratio F:M) | Symptomatic Infections | ||
|---|---|---|---|---|---|---|
| d All Participants (Ratio F:M) | e HFMD (Ratio F:M) | f Herpangina (Ratio F:M) | ||||
| Total Number | 5 (15-2-5-7-3) | 14 (9:5) | 18 (7:11) | 12 (3:9) | 6 (4:2) | |
| Enterovirus A | ||||||
| EV-A71 sub. B5 | 3 (2-0-0-1-2) | 4 (3–5) | N/A | 5 (1:4) | 4 (1:3) | 1 (0:1) |
| EV-A71 sub. C1-like | 1 (0-0-3-0-0) | 6 (5–6) | 1 (1:0) | 2 (2:0) | 1 (1:0) | 1 (1:0) |
| EV-A71 sub. C4 | 1 (3-0-0-0-0) | 4 (3-5) | 2 (1:1) | 1 (0:1) | 1 (0:1) | N/A |
| CVA4 | 3 (2-2-1-0-0) | 4 (3–5) | 2 (1:1) | 3 (2:1) | N/A | 3 (2:1) |
| CVA4/16 | 2 (4-0-0-0-1) | 3 (3–4) | 4 (2:2) | 1 (0:1) | 1 (0:1) | N/A |
| CVA6 | 1 (0-0-0-4-0) | 4 (4–4) | 1 (0:1) | 3 (0:3) | 3 (0:3) | N/A |
| CVA10 | 1 (1-0-0-0-0) | 3 (N/A) | N/A | 1 (0:1) | 1 (0:1) | N/A |
| Enterovirus B | ||||||
| CVA9 | 1 (0-0-1-0-0) | 4 (N/A) | N/A | 1 (1:0) | 1 (1:0) | N/A |
| EchoE30 | 1 (3-0-0-0-0) | 3.5 (3–4) | 2 (2:0) | 1 (1:0) | N/A | 1 (1:0) |
| EchoE11 | 1 (0-0-0-2-0) | 5 (5–5) | 2 (2:0) | N/A | N/A | N/A |
| Event No. | Index Case No. | Enterovirus Genotypes | Blister Swabs c | Quantitative Cycles of RT-PCR of Throat Swabs at Visit | Quantitative Cycles of RT-PCR of Stool Samples at Visit | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 2 | 3 | 4 | 5 | ||||
| 1 | 2 | CVA4 | - | Neg | - | Neg | Neg | Neg | - | - | 22.53 | Neg |
| 2 | 3 | EV-A71 sub. B5 | - | 19.91 a,d | - | 32.97 | 35.20 | Neg | - | - | 32.01 | Neg |
| 3 | 5 | EV-A71 sub. C4 | Neg | 25.08 a | - | - | - | - | - | - | - | 23.54 |
| 4 | 6 | CVA10 | Neg | 25.50 | - | - | - | Neg | - | - | Neg | - |
| 5 | 7 | EV-A71 sub. B5 and EchoE30 b | Neg | 28.70 a,d | - | Neg | 29.73 a,d | - | 24.48 | - | Neg | Neg |
| 6 | 9 | EchoE30 | - | - | - | - | - | - | 22.04 | 34.77 | - | Neg |
| 7 | 4 | CVA4 | - | 28.16 | 29.40 | Neg | 34.50 | - | - | 35.73 | 38.49 | 35.57 |
| 8 | 8 | CVA9 | Neg | Neg | Neg | 24.79 | - | - | - | - | - | - |
| 9 | 11 a | CVA4 | Neg | Neg | - | - | - | - | Neg | Neg | 32.15 | - |
| 10 | 19 | EV-A71 sub. C1-like | 26.02 | - | Neg | Neg | Neg | - | 36.21 | - | - | - |
| 10 | 20 a | EV-A71 sub. C1-like | - | - | 23.89 | Neg | Neg | - | - | 23.36 | 31.17 | - |
| 11 | 1 | EV-A71 sub. B5 | Neg | 29.70 | - | Neg | - | Neg | - | - | - | - |
| 12 | 14 | CVA6 | 35.38 & 20.75 c | 29.11 | - | - | - | - | Neg | - | Neg | 23.02 |
| 12 | 16 | CVA6 | Neg | 30.48 | - | - | - | - | 33.41 | - | - | - |
| 12 | 17 | CVA6 | Neg | 32.04 | - | - | - | - | - | - | - | - |
| 13 | 10 | EV-A71 sub. B5 | Neg | 34.71 | - | - | - | - | 22.58 | 32.55 | Neg | Neg |
| 14 | 13 | EV-A71 sub. B5 | Neg | Neg | - | - | - | - | 25.06 | 28.35 | 29.07 | 30.98 |
| 15 | 15 | CVA4/16 | Neg | 34.58 | - | - | - | - | Neg | Neg | Neg | Neg |
| A total of the virus-detected specimens (%) | 2/13 (15) | 11/15 (73) | 2/4 (50) | 2/8 (25) | 3/6 (50) | 0/4 (0) | 6/9 (67) | 5/7 (71) | 6/11 (55) | 4/10 (40) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sittikul, P.; Batty, E.M.; Yodsawat, P.; Nuanpirom, J.; Kosoltanapiwat, N.; Sangket, U.; Chatchen, S.; Day, N.P.J.; Thaipadungpanit, J. Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020. Viruses 2023, 15, 1397. https://doi.org/10.3390/v15061397
Sittikul P, Batty EM, Yodsawat P, Nuanpirom J, Kosoltanapiwat N, Sangket U, Chatchen S, Day NPJ, Thaipadungpanit J. Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020. Viruses. 2023; 15(6):1397. https://doi.org/10.3390/v15061397
Chicago/Turabian StyleSittikul, Pichamon, Elizabeth M. Batty, Prasert Yodsawat, Jiratchaya Nuanpirom, Nathamon Kosoltanapiwat, Unitsa Sangket, Supawat Chatchen, Nicholas P. J. Day, and Janjira Thaipadungpanit. 2023. "Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020" Viruses 15, no. 6: 1397. https://doi.org/10.3390/v15061397
APA StyleSittikul, P., Batty, E. M., Yodsawat, P., Nuanpirom, J., Kosoltanapiwat, N., Sangket, U., Chatchen, S., Day, N. P. J., & Thaipadungpanit, J. (2023). Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020. Viruses, 15(6), 1397. https://doi.org/10.3390/v15061397