

Polymorphisms Related to Iron Homeostasis Associate with Liver Disease in Chronic Hepatitis C

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. SNP Genotyping
2.3. Gene Expression Analysis
2.4. Statistical Analysis
3. Results
3.1. Histopathological Changes in the Liver
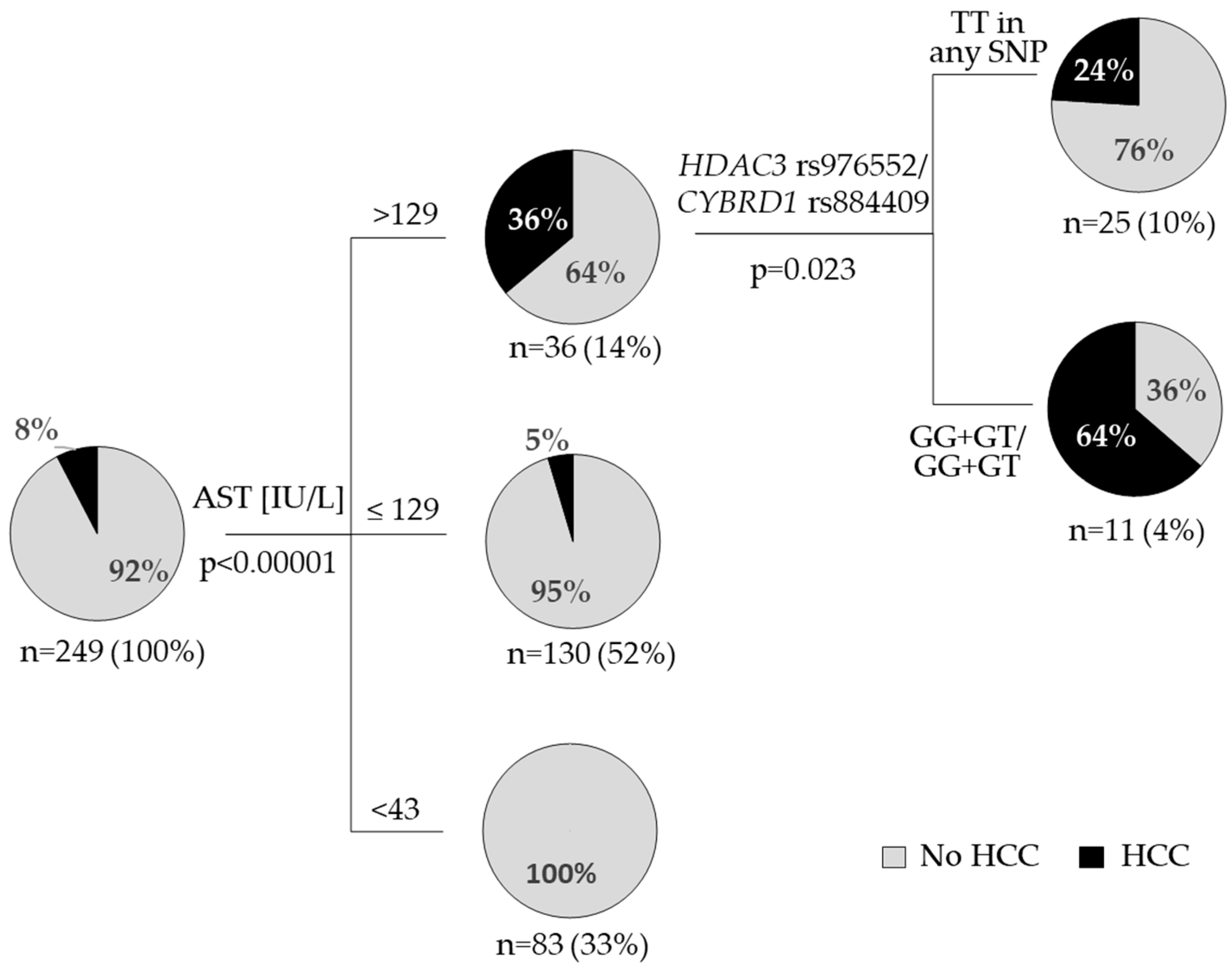
3.2. HCC Occurrence
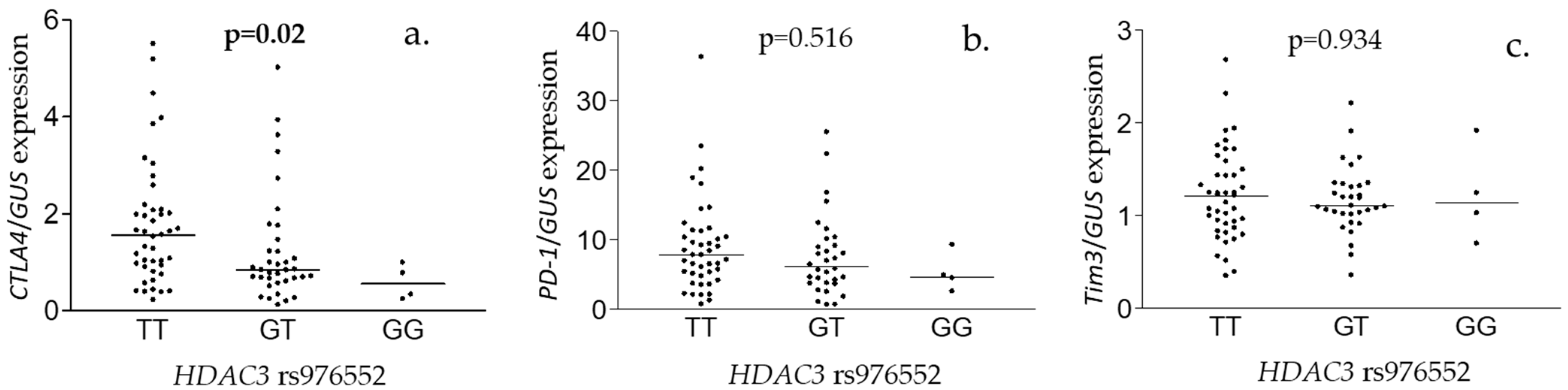
3.3. Hepatic Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N.; EASL HEPAHEALTH Steering Committee. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- World Health Organization. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections, 2021. Accountability for the Global Health Sector Strategies 2016–2021: Actions for Impact; WHO: Geneva, Switzerland, 2021; ISBN 978-92-4-002707-7. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Jühling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 52. [Google Scholar] [CrossRef] [Green Version]
- Kowdley, K.V.; Gochanour, E.M.; Sundaram, V.; Shah, R.A.; Handa, P. Hepcidin Signaling in Health and Disease: Ironing Out the Details. Hepatol. Commun. 2021, 5, 723–735. [Google Scholar] [CrossRef]
- de Campos, W.N.; Massaro, J.D.; Cançado, E.; Wiezel, C.; Simões, A.L.; Teixeira, A.C.; de Souza, F.F.; Mendes-Junior, C.T.; Martinelli, A.; Donadi, E.A. Comprehensive analysis of HFE gene in hereditary hemochromatosis and in diseases associated with acquired iron overload. World J. Hepatol. 2019, 11, 186–198. [Google Scholar] [CrossRef]
- Pichler, I.; Minelli, C.; Sanna, S.; Tanaka, T.; Schwienbacher, C.; Naitza, S.; Porcu, E.; Pattaro, C.; Busonero, F.; Zanon, A.; et al. Identification of a common variant in the TFR2 gene implicated in the physiological regulation of serum iron levels. Hum. Mol. Genet. 2011, 20, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Wahedi, M.; Wortham, A.M.; Kleven, M.D.; Zhao, N.; Jue, S.; Enns, C.A.; Zhang, A.S. Matriptase-2 suppresses hepcidin expression by cleaving multiple components of the hepcidin induction pathway. J. Biol. Chem. 2017, 292, 18354–18371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Fracanzani, A.L.; Rametta, R.; Fraquelli, M.; Soverini, G.; Pelusi, S.; Dongiovanni, P.; Conte, D.; Fargion, S. Effect of the A736V TMPRSS6 polymorphism on the penetrance and clinical expression of hereditary hemochromatosis. J. Hepatol. 2012, 57, 1319–1325. [Google Scholar] [CrossRef]
- Georgopoulou, U.; Dimitriadis, A.; Foka, P.; Karamichali, E.; Mamalaki, A. Hepcidin and the iron enigma in HCV infection. Virulence 2014, 5, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Pulixi, E.A.; Arosio, P.; Cremonesi, L.; Biasiotto, G.; Dongiovanni, P.; Maggioni, M.; Fargion, S.; Fracanzani, A.L. Relative contribution of iron genes, dysmetabolism and hepatitis C virus (HCV) in the pathogenesis of altered iron regulation in HCV chronic hepatitis. Haematologica 2007, 92, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirei, K.; Matsuoka, S.; Nakamura, H.; Matsumura, H.; Moriyama, M. Incidence of hepatocellular carcinoma reduced by phlebotomy treatment in patients with chronic hepatitis C. Intern. Med. 2015, 54, 107–117. [Google Scholar] [CrossRef]
- Kato, J.; Miyanishi, K.; Kobune, M.; Nakamura, T.; Takada, K.; Takimoto, R.; Kawano, Y.; Takahashi, S.; Takahashi, M.; Sato, Y.; et al. Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J. Gastroenterol. 2007, 42, 830–836. [Google Scholar] [CrossRef]
- Dimitriadis, A.; Foka, P.; Kyratzopoulou, E.; Karamichali, E.; Petroulia, S.; Tsitoura, P.; Kakkanas, A.; Eliadis, P.; Georgopoulou, U.; Mamalaki, A. The Hepatitis C virus NS5A and core proteins exert antagonistic effects on HAMP gene expression: The hidden interplay with the MTF-1/MRE pathway. FEBS Open Bio. 2021, 11, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Hasan, Y.; Brown, K. Viral eradication restores normal iron status in chronic hepatitis C patients with abnormal iron studies. Ann. Hepatol. 2020, 19, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L.; Hu, J.H.; Yen, C.H.; Chen, K.H.; Kuo, C.J.; Lin, M.S.; Lee, C.H.; Chen, S.C.; Chien, R.N. Evolution of ferritin levels in hepatitis C patients treated with antivirals. Sci. Rep. 2020, 10, 19744. [Google Scholar] [CrossRef] [PubMed]
- Inomata, S.; Anan, A.; Yamauchi, E.; Yamauchi, R.; Kunimoto, H.; Takata, K.; Tanaka, T.; Yokoyama, K.; Morihara, D.; Takeyama, Y.; et al. Changes in the Serum Hepcidin-to-ferritin Ratio with Erythroferrone after Hepatitis C Virus Eradication Using Direct-acting Antiviral Agents. Intern. Med. 2019, 58, 2915–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wróblewska, A.; Bernat, A.; Woziwodzka, A.; Markiewicz, J.; Romanowski, T.; Bielawski, K.P.; Smiatacz, T.; Sikorska, K. Interferon lambda polymorphisms associate with body iron indices and hepatic expression of interferon-responsive long non-coding RNA in chronic hepatitis C. Clin. Exp. Med. 2017, 17, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Tung, B.Y.; Emond, M.J.; Bronner, M.P.; Raaka, S.D.; Cotler, S.J.; Kowdley, K.V. Hepatitis C, iron status, and disease severity: Relationship with HFE mutations. Gastroenterology 2003, 124, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.; Woziwodzka, A.; Wróblewska, A.; Rybicka, M.; Bielawski, K.P.; Sikorska, K.; Bernat, A. Analysis of polymorphism and hepatic expression of duodenal cytochrome b in chronic hepatitis C. J. Gastroenterol. Hepatol. 2017, 32, 482–486. [Google Scholar] [CrossRef]
- Sikorska, K.; Romanowski, T.; Stalke, P.; Izycka Swieszewska, E.; Bielawski, K.P. Association of hepcidin mRNA expression with hepatocyte iron accumulation and effects of antiviral therapy in chronic hepatitis C infection. Hepat. Mon. 2014, 14, e21184. [Google Scholar] [CrossRef] [Green Version]
- Sikorska, K.; Bielawski, K.P.; Stalke, P.; Lakomy, E.A.; Michalska, Z.; Witczak-Malinowska, K.; Romanowski, T. HFE gene mutations in Polish patients with disturbances of iron metabolism: An initial assessment. Int. J. Mol. Med. 2005, 16, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Schemper, M.; Smith, T.L. A note on quantifying follow-up in studies of failure time. Control Clin. Trials 1996, 17, 343–346. [Google Scholar] [CrossRef]
- Halota, W.; Flisiak, R.; Juszczyk, J.; Małkowski, P.; Pawłowska, M.; Simon, K.; Tomasiewicz, K. Recommendations of the Polish Group of Experts for HCV for the treatment of hepatitis C in 2020. Clin. Exp. Hepatol. 2020, 6, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, M.; Woziwodzka, A.; Romanowski, T.; Sznarkowska, A.; Stalke, P.; Dręczewski, M.; Bielawski, K.P. Host genetic background affects the course of infection and treatment response in patients with chronic hepatitis B. J. Clin. Virol. 2019, 120, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, T.R.; Rodriguez, S.; Zapata, C.; Day, I.N. MIDAS: Software for analysis and visualisation of interallelic disequilibrium between multiallelic markers. BMC Bioinform. 2006, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Agoro, R.; Mura, C. Inflammation-induced up-regulation of hepcidin and down-regulation of ferroportin transcription are dependent on macrophage polarization. Blood Cells Mol. Dis. 2016, 61, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yuan, Y.; Kuang, Y.; Li, X. Iron Metabolism and Immune Regulation. Front. Immunol. 2022, 13, 816282. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, K.; Stalke, P.; Izycka-Swieszewska, E.; Romanowski, T.; Bielawski, K.P. The role of iron overload and HFE gene mutations in the era of pegylated interferon and ribavirin treatment of chronic hepatitis C. Med. Sci. Monit. 2010, 16, CR137–CR143. [Google Scholar]
- Höhler, T.; Leininger, S.; Köhler, H.H.; Schirmacher, P.; Galle, P.R. Heterozygosity for the hemochromatosis gene in liver diseases--prevalence and effects on liver histology. Liver 2000, 20, 482–486. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.R.; Lim, P.J.; Duarte, T.L.; Casu, C.; Oosterhuis, D.; Mleczko-Sanecka, K.; Suciu, M.; Da Silva, A.R.; Al-Hourani, K.; Arezes, J.; et al. Hepcidin is regulated by promoter-associated histone acetylation and HDAC3. Nat. Commun. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Miller, R.A.; Patel, R.T.; Chen, J.; Dhir, R.; Wang, H.; Zhang, D.; Graham, M.J.; Unterman, T.G.; Shulman, G.I.; et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012, 18, 934–942. [Google Scholar] [CrossRef]
- López-Rodríguez, R.; Hernández-Bartolomé, Á.; Borque, M.J.; Rodríguez-Muñoz, Y.; Martín-Vílchez, S.; Trapero-Marugán, M.; García-Buey, L.; Muñoz de Rueda, P.; Rodrigo, L.; Vidal-Castiñeira, J.R.; et al. Polymorphisms in histone deacetylases improve the predictive value of IL-28B for chronic hepatitis C therapy. Genes. Immun. 2013, 14, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Ler, S.Y.; Leung, C.H.; Khin, L.W.; Lu, G.D.; Salto-Tellez, M.; Hartman, M.; Iau, P.T.; Yap, C.T.; Hooi, S.C. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population. Oncol. Rep. 2015, 34, 2238–2250. [Google Scholar] [CrossRef] [Green Version]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [Green Version]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Georgakopoulou, V.E.; Sarantis, P.; Antoniou, E.A.; Karamouzis, M.V.; Nonni, A.; Schizas, D.; Diamantis, E.; et al. Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities. J. Pers. Med. 2021, 11, 223. [Google Scholar] [CrossRef]
- Constantine, C.C.; Anderson, G.J.; Vulpe, C.D.; McLaren, C.E.; Bahlo, M.; Yeap, H.L.; Gertig, D.M.; Osborne, N.J.; Bertalli, N.A.; Beckman, K.B.; et al. A novel association between a SNP in CYBRD1 and serum ferritin levels in a cohort study of HFE hereditary haemochromatosis. Br. J. Haematol. 2009, 147, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelucchi, S.; Mariani, R.; Calza, S.; Fracanzani, A.L.; Modignani, G.L.; Bertola, F.; Busti, F.; Trombini, P.; Fraquelli, M.; Forni, G.L.; et al. CYBRD1 as a modifier gene that modulates iron phenotype in HFE p.C282Y homozygous patients. Haematologica 2012, 97, 1818–1825. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Huang, W.; Zhu, C.; Sun, X.; Zhang, Q.; Zhang, L.; Qi, Q.; Bai, X.; Feng, Y.; Wang, C. miR-423-3p activates FAK signaling pathway to drive EMT process and tumor growth in lung adenocarcinoma through targeting CYBRD1. J. Clin. Lab. Anal. 2021, 35, e24044. [Google Scholar] [CrossRef]
- Lemler, D.J.; Lynch, M.L.; Tesfay, L.; Deng, Z.; Paul, B.T.; Wang, X.; Hegde, P.; Manz, D.H.; Torti, S.V.; Torti, F.M. DCYTB is a predictor of outcome in breast cancer that functions via iron-independent mechanisms. Breast Cancer Res. 2017, 19, 25. [Google Scholar] [CrossRef]
- Raziorrouh, B.; Ulsenheimer, A.; Schraut, W.; Heeg, M.; Kurktschiev, P.; Zachoval, R.; Jung, M.C.; Thimme, R.; Neumann-Haefelin, C.; Horster, S.; et al. Inhibitory molecules that regulate expansion and restoration of HCV-specific CD4+ T cells in patients with chronic infection. Gastroenterology 2011, 141, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, e1–e2. [Google Scholar] [CrossRef] [Green Version]
- McMahan, R.H.; Golden-Mason, L.; Nishimura, M.I.; McMahon, B.J.; Kemper, M.; Allen, T.M.; Gretch, D.R.; Rosen, H.R. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J. Clin. Invest. 2010, 120, 4546–4557. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Rumpret, M.; Drylewicz, J.; Ackermans, L.J.E.; Borghans, J.A.M.; Medzhitov, R.; Meyaard, L. Functional categories of immune inhibitory receptors. Nat. Rev. Immunol. 2020, 20, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kang, H.; Lee, H.H.; Kim, C.W. Programmed Cell Death 1 (PD-1) and Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) in Viral Hepatitis. Int. J. Mol. Sci. 2017, 18, 1517. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genotype | Steatosis | Advanced Fibrosis * | Iron Deposits | ||||
|---|---|---|---|---|---|---|---|
| OR (CI 95%) | p | OR (CI 95%) | p | OR (CI 95%) | p | ||
| CYBRD1 rs884409 | TT (n = 140) | 0.5 (0.3–0.9) | 0.029 | 0.7 (0.4–1.4) | NS | 1.0 (0.6–1.9) | NS |
| GG (n = 12) | 2.0 (0.5–7.6) | NS | 3.7 (17.8–0.8) | NS | 1.0 (0.3–3.6) | NS | |
| HDAC5 rs368328 | AA (n = 96) | 2.3 (1.3–4.1) | 0.006 | 2.2 (1.2–4.1) | 0.010 | 1.1 (0.6–2.0) | NS |
| GG (n = 26) | 0.5 (0.2–1.1) | NS | 0.6 (0.3–1.3) | NS | 0.8 (0.3–1.9) | NS | |
| TFR2 rs7385804 | AA (n = 58) | 1.0 (0.5–1.8) | NS | 0.6 (0.3–1.2) | NS | 0.9 (0.4–1.8) | NS |
| CC (n = 42) | 0.8 (0.4–1.6) | NS | 2.4 (1.1–5.3) | 0.035 | 0.6 (0.3–1.2) | NS | |
| TMPRSS6 rs855791 | CC (n = 83) | 0.8 (0.5–1.4) | NS | 0.5 (0.3–0.8) | 0.011 | 1.5 (0.8–2.7) | NS |
| TT (n = 20) | 0.9 (0.4–2.4) | NS | 3.6 (1.1–11.8) | NS | 0.5 (0.2–1.4) | NS | |
| HFE H63D rs1799945 | CC (n = 144) | 0.8 (0.4–1.4) | NS | 0.7 (0.4–1.2) | NS | 0.5 (0.2–0.9) | 0.022 |
| GG (n = 6) | 0.6 (0.1–3.0) | NS | 0.5 (0.1–2.0) | NS | 0.7 (0.1–3.7) | NS | |
| Characteristisc (n = 249) | HCC | p | OR (CI 95%) | ||
|---|---|---|---|---|---|
| No (n = 230) | Yes (n = 19) | ||||
| Age [yr] | 47 ± 1 | 53 ± 2 | 0.022 | ||
| Sex (Male/Female) | 140/90 | 13/6 | 0.686 | ||
| HGB [g/dL] | 14.8 ± 0.1 | 14.6 ± 0.4 | 0.579 | ||
| ALT [IU/L] | 109 ± 6 | 187 ± 23 | 0.0002 | ||
| AST [IU/L] | 72 ± 3 | 164 ± 19 | <0.00001 | ||
| GGT [IU/L] | 97 ± 6 | 152 ± 20 | 0.002 | ||
| Bilirubin [mg/dL] | 0.8 ± 0.04 | 1.2 ± 0.1 | 0.0006 | ||
| sFe [μg/dL] | 147 ± 4 | 215 ± 15 | 0.00004 | ||
| Transferrin saturation [%] | 42 ± 1 | 67 ± 10 | 0.010 | ||
| sFerritin [ng/mL] | 351 ± 31 | 609 ± 89 | 0.002 | ||
| Histopathology n = 211 | No (n = 97) | Yes (n = 14) | p | ||
| Inflammation grade | 2 (2/2) | 3 (2/3) | 0.001 | ||
| Fibrosis grade | 2 (1/3) | 3 (2/3) | 0.276 | ||
| Iron deposits grade (0–3) | 0 (0/1) | 0 (0/1) | 0.618 | ||
| Steatosis grade (0–3) | 1 (0/2) | 1 (0/2) | 0.719 | ||
| Hepatocyte iron deposits present (yes/no) | 70/127 | 6/8 | 0.621 | ||
| Hepatocyte steatosis present (yes/no) | 118/79 | 8/6 | 0.937 | ||
| Liver fibrosis present (yes/no) | 119/78 | 12/2 | 0.050 | ||
| Polymorphism n = 249 | No (n = 230) | Yes (n = 19) | p | ||
| HDAC3 rs976552 | TT | 155 (96%) | 7 (4%) | 0.026 # | |
| GT | 70 (86%) | 11 (14%) | |||
| GG | 5 (83%) | 1 (17%) | |||
| TT | 155 (96%) | 7 (4%) | 0.011 * | 3.6 (1.3–9.8) | |
| GG + GT | 75 (86%) | 12 (14%) | |||
| CYBRD1 rs884409 | TT | 155 (95%) | 8 (5%) | 0.041 * | 2.5 (1.0–7.1) |
| GT + GG | 75 (87%) | 11 (13%) | |||
| HDAC3 rs976552/ CYBRD1 rs884409 | TT in any SNP | 203 (95%) | 10 (5%) | 0.001 * | 8.1 (2.2–29.2) |
| GG + GT/GG + GT | 27 (75%) | 9 (25%) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróblewska, A.; Woziwodzka, A.; Rybicka, M.; Bielawski, K.P.; Sikorska, K. Polymorphisms Related to Iron Homeostasis Associate with Liver Disease in Chronic Hepatitis C. Viruses 2023, 15, 1710. https://doi.org/10.3390/v15081710
Wróblewska A, Woziwodzka A, Rybicka M, Bielawski KP, Sikorska K. Polymorphisms Related to Iron Homeostasis Associate with Liver Disease in Chronic Hepatitis C. Viruses. 2023; 15(8):1710. https://doi.org/10.3390/v15081710
Chicago/Turabian StyleWróblewska, Anna, Anna Woziwodzka, Magda Rybicka, Krzysztof P. Bielawski, and Katarzyna Sikorska. 2023. "Polymorphisms Related to Iron Homeostasis Associate with Liver Disease in Chronic Hepatitis C" Viruses 15, no. 8: 1710. https://doi.org/10.3390/v15081710
APA StyleWróblewska, A., Woziwodzka, A., Rybicka, M., Bielawski, K. P., & Sikorska, K. (2023). Polymorphisms Related to Iron Homeostasis Associate with Liver Disease in Chronic Hepatitis C. Viruses, 15(8), 1710. https://doi.org/10.3390/v15081710