

Evolution of the SARS-CoV-2 Omicron Variants: Genetic Impact on Viral Fitness

 and
and 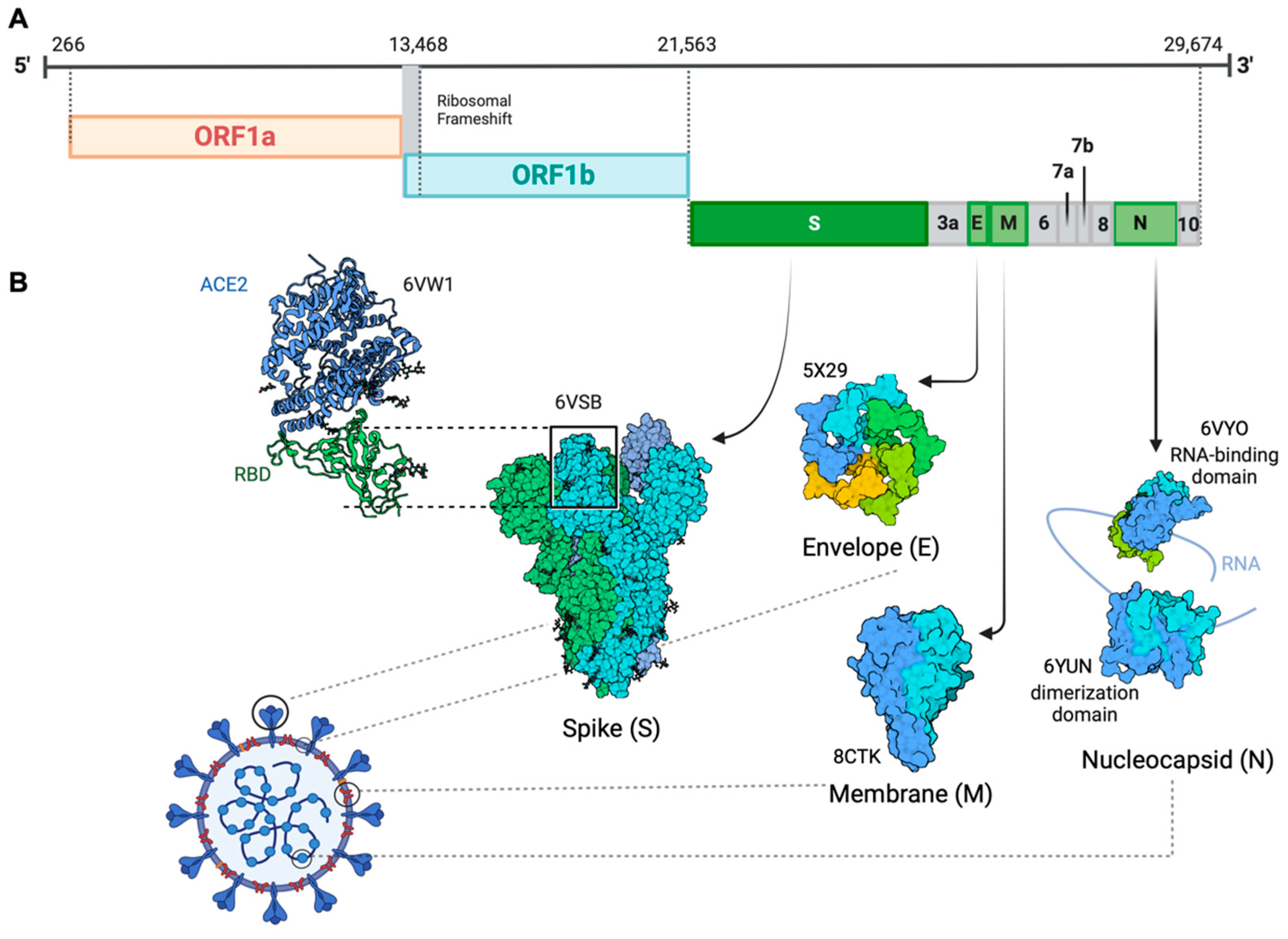{kind=link}
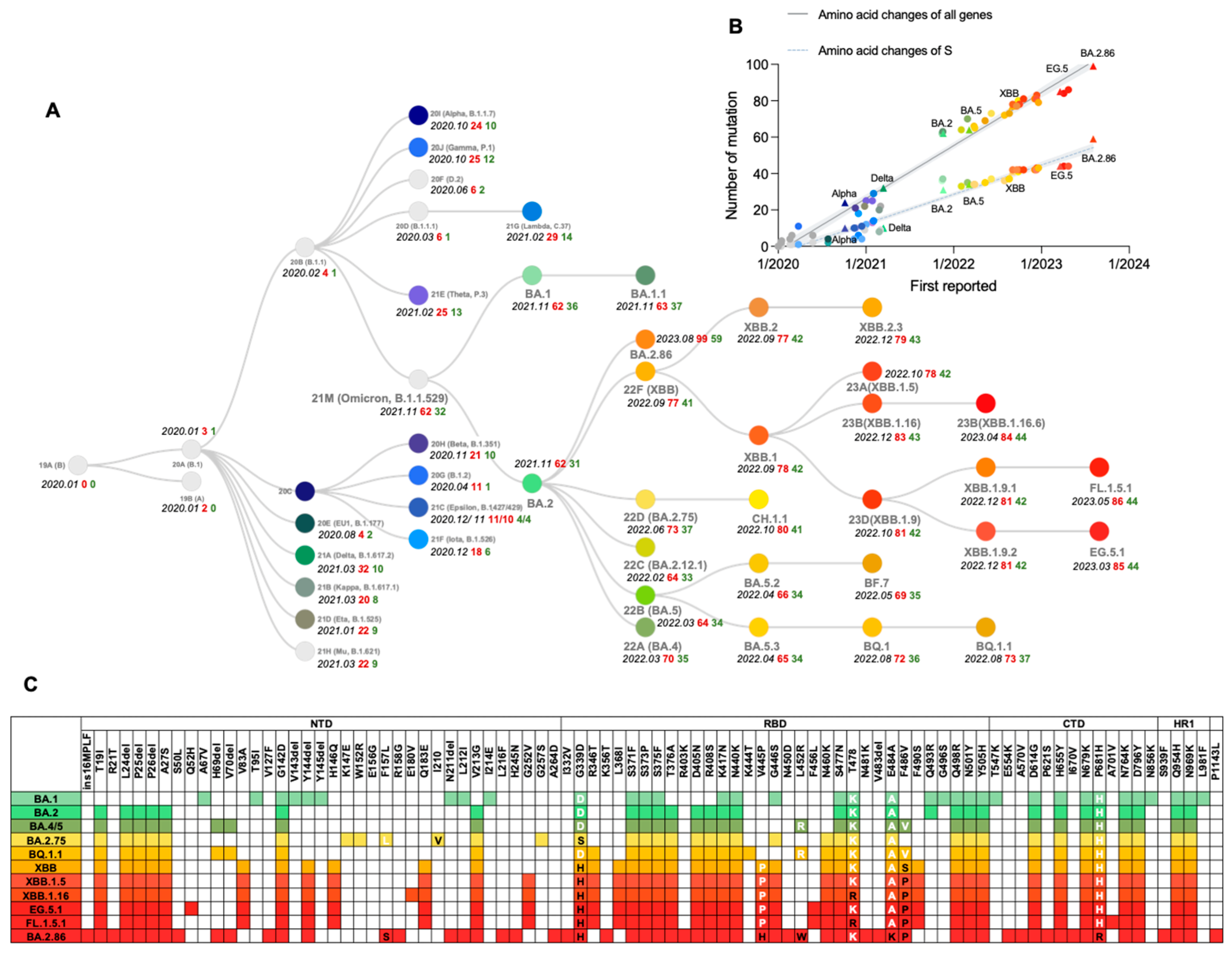{kind=link}
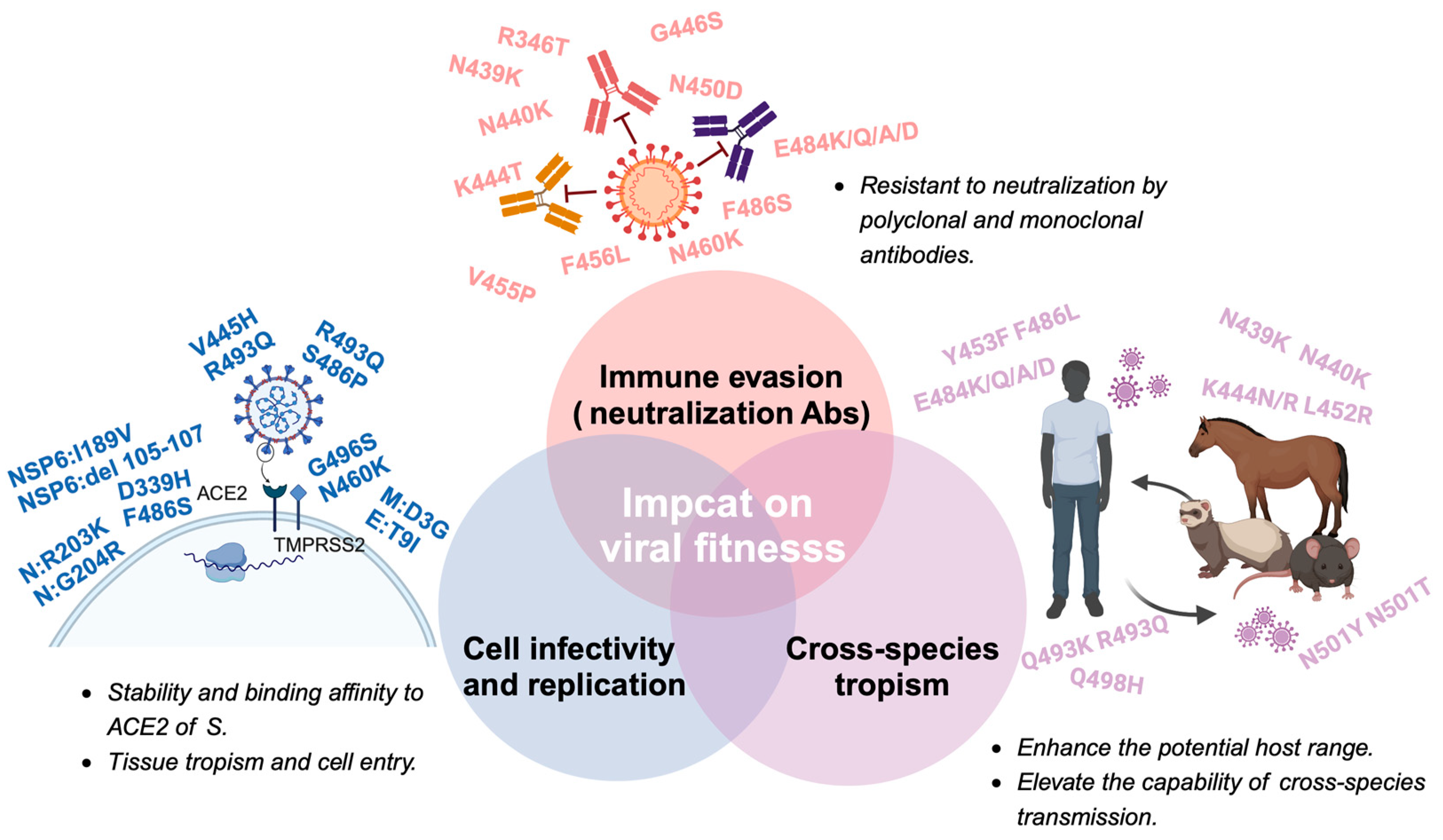{kind=link}
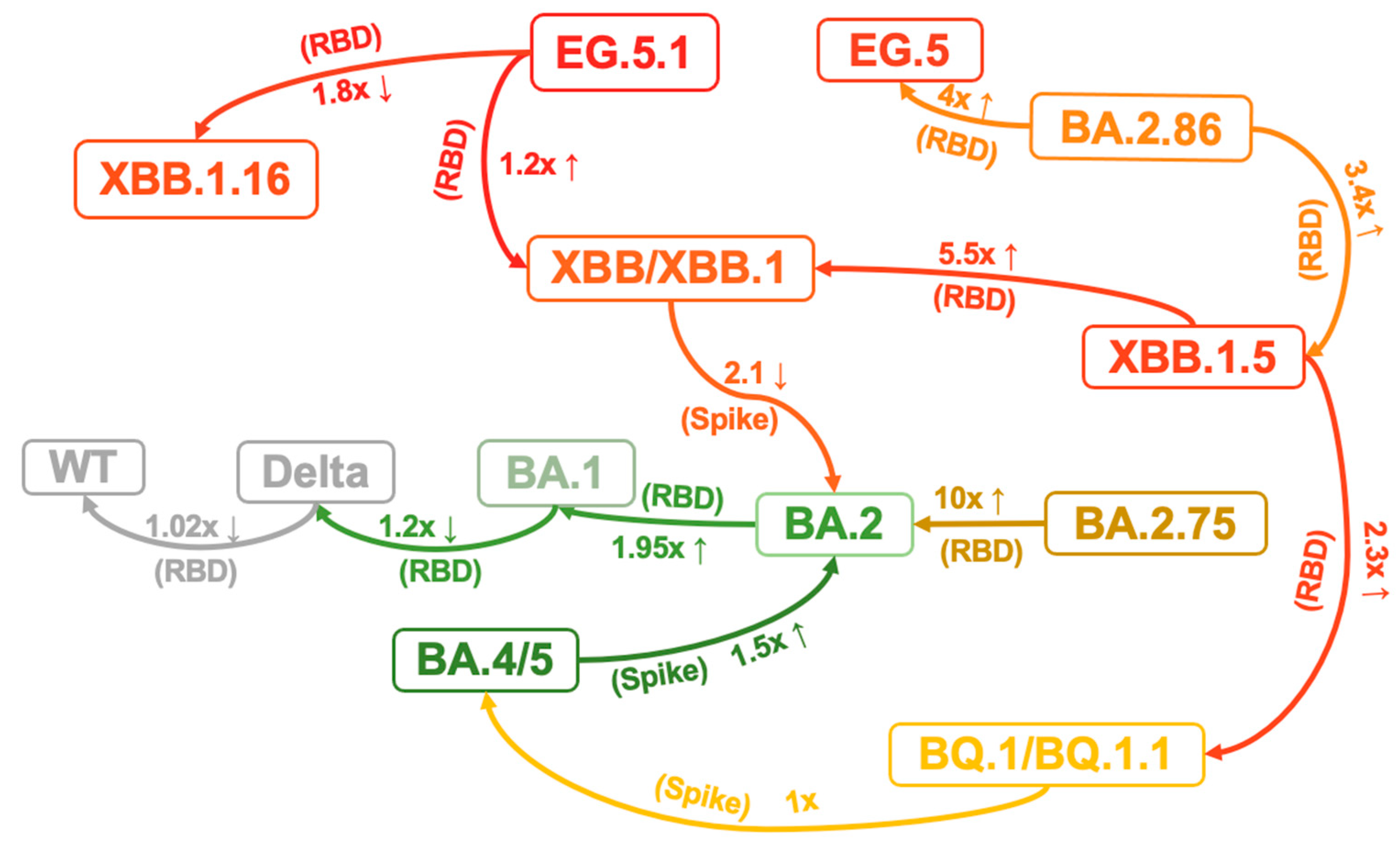{kind=link}
Abstract
:1. Molecular Biological Characteristics of SARS-CoV-2
2. The Evolution of SARS-CoV-2 Variants
3. Hypotheses about the Origin of Omicron
4. Impacts of Mutations in Omicron on Viral Fitness
4.1. Immune Evasion
4.1.1. Neutralization by Immune Sera
4.1.2. Neutralization by Monoclonal Antibodies
4.2. Cell Infectivity and Replication
4.2.1. Stability and Binding Affinity to ACE2 of S Protein
4.2.2. Tissue Tropism and Cell Entry
4.3. The Impact of Mutations beyond S Protein
4.4. Cross-Species Transmission
5. Strategies for Preventing Emerging Variants
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alsobaie, S. Understanding the Molecular Biology of SARS-CoV-2 and the COVID-19 Pandemic: A Review. Infect. Drug Resist. 2021, 14, 2259–2268. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A Structural Analysis of M Protein in Coronavirus Assembly and Morphology. J. Struct. Biol. 2011, 174, 11. [Google Scholar] [CrossRef]
- Nal, B.; Chan, C.; Kien, F.; Siu, L.; Tse, J.; Chu, K.; Kam, J.; Staropoli, I.; Crescenzo-Chaigne, B.; Escriou, N.; et al. Differential Maturation and Subcellular Localization of Severe Acute Respiratory Syndrome Coronavirus Surface Proteins S, M and E. J. Gen. Virol. 2005, 86, 1423–1434. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A Severe Acute Respiratory Syndrome Coronavirus That Lacks the E Gene Is Attenuated In Vitro and In Vivo. J. Virol. 2007, 81, 1701. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- Masters, P.S.; Sturman, L.S. Background Paper. Functions of the Coronavirus Nucleocapsid Protein. Adv. Exp. Med. Biol. 1990, 276, 235–238. [Google Scholar] [CrossRef]
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef]
- Chang, C.; Sue, S.-C.; Yu, T.; Hsieh, C.-M.; Tsai, C.-K.; Chiang, Y.-C.; Lee, S.; Hsiao, H.; Wu, W.-J.; Chang, W.-L.; et al. Modular Organization of SARS Coronavirus Nucleocapsid Protein. J. Biomed. Sci. 2006, 13, 59. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Identification of In Vivo-Interacting Domains of the Murine Coronavirus Nucleocapsid Protein. J. Virol. 2009, 83, 7221. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.M.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Li, F.; Berardi, M.; Li, W.; Farzan, M.; Dormitzer, P.R.; Harrison, S.C. Conformational States of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Ectodomain. J. Virol. 2006, 80, 6794–6800. [Google Scholar] [CrossRef]
- Li, F. Evidence for a Common Evolutionary Origin of Coronavirus Spike Protein Receptor-Binding Subunits. J. Virol. 2012, 86, 2856–2858. [Google Scholar] [CrossRef]
- Li, F. Receptor Recognition and Cross-Species Infections of SARS Coronavirus. Antivir. Res. 2013, 100, 246–254. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A Pan-Coronavirus Fusion Inhibitor Targeting the HR1 Domain of Human Coronavirus Spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-nCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and Bat RaTG13 Spike Glycoprotein Structures Inform on Virus Evolution and Furin Cleavage Effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef] [PubMed]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. Natural Selection in the Evolution of SARS-CoV-2 in Bats Created a Generalist Virus and Highly Capable Human Pathogen. PLoS Biol. 2021, 19, e3001115. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Mohsin, M.; Mahmud, S. Omicron SARS-CoV-2 Variant of Concern. Medicine 2022, 101, e29165. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Coutinho, R.M.; Marquitti, F.M.D.; Ferreira, L.S.; Borges, M.E.; da Silva, R.L.P.; Canton, O.; Portella, T.P.; Poloni, S.; Franco, C.; Plucinski, M.M.; et al. Model-Based Estimation of Transmissibility and Reinfection of SARS-CoV-2 P.1 Variant. Commun. Med. 2021, 1, 48. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of Mortality in Patients Infected with SARS-CoV-2 Variant of Concern 202012/1: Matched Cohort Study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Liu, Y.; Rocklöv, J. The Reproductive Number of the Delta Variant of SARS-CoV-2 Is Far Higher Compared to the Ancestral SARS-CoV-2 Virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gayle, A.A.; Wilder-Smith, A.; Rocklöv, J. The Reproductive Number of COVID-19 Is Higher Compared to SARS Coronavirus. J. Travel Med. 2020, 27, taaa021. [Google Scholar] [CrossRef]
- Earnest, R.; Uddin, R.; Matluk, N.; Renzette, N.; Turbett, S.E.; Siddle, K.J.; Loreth, C.; Adams, G.; Tomkins-Tinch, C.H.; Petrone, M.E.; et al. Comparative Transmissibility of SARS-CoV-2 Variants Delta and Alpha in New England, USA. Cell Rep. Med. 2022, 3, 100583. [Google Scholar] [CrossRef]
- Liu, Y.; Rocklöv, J. The Effective Reproductive Number of the Omicron Variant of SARS-CoV-2 Is Several Times Relative to Delta. J. Travel Med. 2022, 29, taac037. [Google Scholar] [CrossRef]
- Du, Z.; Hong, H.; Wang, S.; Ma, L.; Liu, C.; Bai, Y.; Adam, D.C.; Tian, L.; Wang, L.; Lau, E.H.Y.; et al. Reproduction Number of the Omicron Variant Triples That of the Delta Variant. Viruses 2022, 14, 821. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 Omicron Variants BA.1 to BA.5: Implications for Immune Escape and Transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Hu, B.; Chai, Y.; Shuai, H.; Liu, H.; Shi, J.; Liu, Y.; Yoon, C.; Zhang, J.; Hu, J.-C.; et al. Virological Features and Pathogenicity of SARS-CoV-2 Omicron BA.2. Cell Rep. Med. 2022, 3, 100743. [Google Scholar] [CrossRef]
- Lino, A.; Cardoso, M.A.; Martins-Lopes, P.; Gonçalves, H.M.R. Omicron–The New SARS-CoV-2 Challenge? Rev. Med. Virol. 2022, 32, e2358. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2 Subvariants, Including BA.4 and BA.5. Cell 2022, 185, 3992–4007.e16. [Google Scholar] [CrossRef]
- Sabbatucci, M.; Vitiello, A.; Clemente, S.; Zovi, A.; Boccellino, M.; Ferrara, F.; Cimmino, C.; Langella, R.; Ponzo, A.; Stefanelli, P.; et al. Omicron Variant Evolution on Vaccines and Monoclonal Antibodies. Inflammopharmacology 2023, 31, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Zappa, M.; Verdecchia, P.; Angeli, F. Knowing the New Omicron BA.2.75 Variant (‘Centaurus’): A Simulation Study. Eur. J. Intern. Med. 2022, 105, 107–108. [Google Scholar] [CrossRef]
- Saito, A.; Tamura, T.; Zahradnik, J.; Deguchi, S.; Tabata, K.; Anraku, Y.; Kimura, I.; Ito, J.; Yamasoba, D.; Nasser, H.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2.75 Variant. Cell Host Microbe 2022, 30, 1540–1555.e15. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Ito, J.; Suzuki, R.; Uriu, K.; Itakura, Y.; Zahradnik, J.; Kimura, K.T.; Deguchi, S.; Wang, L.; Lytras, S.; Tamura, T.; et al. Convergent Evolution of SARS-CoV-2 Omicron Subvariants Leading to the Emergence of BQ.1.1 Variant. Nat. Commun. 2023, 14, 2671. [Google Scholar] [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 Binding and Antibody Evasion in Enhanced Transmissibility of XBB.1.5. Lancet Infect. Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 27 September 2023).
- Yang, S.; Yu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; Wang, J.; Yu, L.; et al. Antigenicity and Infectivity Characterisation of SARS-CoV-2 BA.2.86. Lancet Infect. Dis. 2023, 23, e457–e459. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, W.; Dong, W.; Xu, J. Origin and Evolutionary Analysis of the SARS-CoV-2 Omicron Variant. J. Biosaf. Biosecurity 2022, 4, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Herrera-Carrillo, E. SARS-CoV-2 Evolution: On the Sudden Appearance of the Omicron Variant. J. Virol. 2022, 96, e0009022. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Shan, K.-J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a Mouse Origin of the SARS-CoV-2 Omicron Variant. J. Genet. Genom. 2021, 48, 1111–1121. [Google Scholar] [CrossRef]
- Where Did ‘Weird’ Omicron Come from? Available online: https://www.science.org/content/article/where-did-weird-omicron-come (accessed on 21 September 2023).
- Where Did Omicron Come from? Three Key Theories. Available online: https://www.nature.com/articles/d41586-022-00215-2 (accessed on 11 August 2023).
- Wargo, A.R.; Kurath, G. Viral Fitness: Definitions, Measurement, and Current Insights. Curr. Opin. Virol. 2012, 2, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, M.; Wu, Y.; Wang, J.; Hong, Y.; Huang, Y.; Yuan, L.; Ma, J.; Wang, K.; Wang, S.; et al. Cross-Species Tropism and Antigenic Landscapes of Circulating SARS-CoV-2 Variants. Cell Rep. 2022, 38, 110558. [Google Scholar] [CrossRef]
- Chen, S.; Huang, Z.; Guo, Y.; Guo, H.; Jian, L.; Xiao, J.; Yao, X.; Yu, H.; Cheng, T.; Zhang, Y.; et al. Evolving Spike Mutations in SARS-CoV-2 Omicron Variants Facilitate Evasion from Breakthrough Infection-Acquired Antibodies. Cell Discov. 2023, 9, 86. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.-W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking Antibody Evasion Manifested by the Omicron Variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Q.; Liang, Z.; Li, T.; Liu, S.; Cui, Q.; Nie, J.; Wu, Q.; Qu, X.; Huang, W.; et al. The Significant Immune Escape of Pseudotyped SARS-CoV-2 Variant Omicron. Emerg. Microbes Infect. 2022, 11, 1–5. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Zhang, R.M.; Ho, J.; Mohri, H.; Valdez, R.; Manthei, D.M.; Gordon, A.; Liu, L.; Ho, D.D. Antibody Neutralisation of Emerging SARS-CoV-2 Subvariants: EG.5.1 and XBC.1.6. Lancet Infect. Dis. 2023, 23, e397–e398. [Google Scholar] [CrossRef]
- Uriu, K.; Ito, J.; Kosugi, Y.; Tanaka, Y.L.; Mugita, Y.; Guo, Z.; Hinay, A.A.; Putri, O.; Kim, Y.; Shimizu, R.; et al. Transmissibility, Infectivity, and Immune Evasion of the SARS-CoV-2 BA.2.86 Variant. Lancet Infect. Dis. 2023, 23, e460–e461. [Google Scholar] [CrossRef]
- Khan, K.; Lustig, G.; Römer, C.; Reedoy, K.; Jule, Z.; Karim, F.; Ganga, Y.; Bernstein, M.; Baig, Z.; Jackson, L.; et al. Evolution and Neutralization Escape of the SARS-CoV-2 BA.2.86 Subvariant. Nat. Commun. 2023, 14, 8078. [Google Scholar] [CrossRef]
- Willett, B.J.; Logan, N.; Scott, S.; Davis, C.; McSorley, T.; Asamaphan, P.; Hosie, M.J.; Olmo, P.; Grove, J.; Orton, R.; et al. Omicron BA.2.86 Cross-Neutralising Activity in Community Sera from the UK. Lancet 2023, 402, 2075–2076. [Google Scholar] [CrossRef]
- ACTIV-3/Therapeutics for Inpatients with COVID-19 (TICO) Study Group. Efficacy and Safety of Two Neutralising Monoclonal Antibody Therapies, Sotrovimab and BRII-196 plus BRII-198, for Adults Hospitalised with COVID-19 (TICO): A Randomised Controlled Trial. Lancet Infect. Dis. 2022, 22, 622–635. [Google Scholar] [CrossRef]
- Evering, T.H.; Chew, K.W.; Giganti, M.J.; Moser, C.; Pinilla, M.; Wohl, D.A.; Currier, J.S.; Eron, J.J.; Javan, A.C.; Bender Ignacio, R.; et al. Safety and Efficacy of Combination SARS-CoV-2 Neutralizing Monoclonal Antibodies Amubarvimab Plus Romlusevimab in Nonhospitalized Patients With COVID-19. Ann. Intern. Med. 2023, 176, 658–666. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Y.; Zheng, R.; Si, S.; Xi, Y.; Deng, X.; Wang, G.; Zhou, L.; Li, M.; Wang, Y.; et al. Effect of the Timing of Amubarvimab/Romlusevimab (BRII-196/198) Administration on Progression to Severe Disease in Elderly Patients with COVID-19 Infection: A Retrospective Cohort Study. Intensive Care Res. 2023, 3, 103–111. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; COVID-19 Genomics UK (COG-UK) Consortium; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; et al. SARS-CoV-2 Variant Evasion of Monoclonal Antibodies Based on in Vitro Studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Gruell, H.; Vanshylla, K.; Tober-Lau, P.; Hillus, D.; Schommers, P.; Lehmann, C.; Kurth, F.; Sander, L.E.; Klein, F. mRNA Booster Immunization Elicits Potent Neutralizing Serum Activity against the SARS-CoV-2 Omicron Variant. Nat. Med. 2022, 28, 477–480. [Google Scholar] [CrossRef]
- Tang, Z.; Yu, P.; Guo, Q.; Chen, M.; Lei, Y.; Zhou, L.; Mai, W.; Chen, L.; Deng, M.; Kong, W.; et al. Clinical Characteristics and Host Immunity Responses of SARS-CoV-2 Omicron Variant BA.2 with Deletion of ORF7a, ORF7b and ORF8. Virol. J. 2023, 20, 106. [Google Scholar] [CrossRef]
- Bebtelovimab. Available online: https://go.drugbank.com/drugs/DB16755 (accessed on 24 December 2023).
- Yamasoba, D.; Kosugi, Y.; Kimura, I.; Fujita, S.; Uriu, K.; Ito, J.; Sato, K. Neutralisation Sensitivity of SARS-CoV-2 Omicron Subvariants to Therapeutic Monoclonal Antibodies. Lancet Infect. Dis. 2022, 22, 942–943. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Y.; Wang, Y.; Zhang, C.; Hong, Q.; Gu, C.; Xu, R.; Wang, T.; Yang, Y.; Zang, J.; et al. Mapping Cross-Variant Neutralizing Sites on the SARS-CoV-2 Spike Protein. Emerg. Microbes Infect. 2022, 11, 351–367. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Zhang, Z.; Yisimayi, A.; Hao, X.; Bao, L.; Yuan, F.; Yu, Y.; Du, S.; Wang, J.; et al. Rational Identification of Potent and Broad Sarbecovirus-Neutralizing Antibody Cocktails from SARS Convalescents. Cell Rep. 2022, 41, 111845. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Casner, R.G.; Guo, Y.; Wang, Q.; Iketani, S.; Chan, J.F.-W.; Yu, J.; Dadonaite, B.; Nair, M.S.; Mohri, H.; et al. Antibodies That Neutralize All Current SARS-CoV-2 Variants of Concern by Conformational Locking. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular Basis of Receptor Binding and Antibody Neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.H.L. Transmissibility and Transmission of Respiratory Viruses. Nat. Rev. Microbiol. 2021, 19, 528–545. [Google Scholar] [CrossRef]
- Minhaz Ud-Dean, S.M. Structural Explanation for the Effect of Humidity on Persistence of Airborne Virus: Seasonality of Influenza. J. Theor. Biol. 2010, 264, 822–829. [Google Scholar] [CrossRef]
- Ausar, S.F.; Rexroad, J.; Frolov, V.G.; Look, J.L.; Konar, N.; Middaugh, C.R. Analysis of the Thermal and pH Stability of Human Respiratory Syncytial Virus. Mol. Pharm. 2005, 2, 491–499. [Google Scholar] [CrossRef]
- Ijaz, M.K.; Brunner, A.H.; Sattar, S.A.; Nair, R.C.; Johnson-Lussenburg, C.M. Survival Characteristics of Airborne Human Coronavirus 229E. J. Gen. Virol. 1985, 66, 2743–2748. [Google Scholar] [CrossRef]
- Bajimaya, S.; Frankl, T.; Hayashi, T.; Takimoto, T. Cholesterol Is Required for Stability and Infectivity of Influenza A and Respiratory Syncytial Viruses. Virology 2017, 510, 234–241. [Google Scholar] [CrossRef]
- Mateu, M.G. Assembly, Stability and Dynamics of Virus Capsids. Arch. Biochem. Biophys. 2013, 531, 65–79. [Google Scholar] [CrossRef]
- Saha, B.; Wong, C.M.; Parks, R.J. The Adenovirus Genome Contributes to the Structural Stability of the Virion. Viruses 2014, 6, 3563–3583. [Google Scholar] [CrossRef]
- Gerba, C.P.; Betancourt, W.Q. Viral Aggregation: Impact on Virus Behavior in the Environment. Environ. Sci. Technol. 2017, 51, 7318–7325. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural Basis of Human ACE2 Higher Binding Affinity to Currently Circulating Omicron SARS-CoV-2 Sub-Variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Qu, P.; Xu, K.; Faraone, J.N.; Goodarzi, N.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; Saif, L.J.; et al. Immune Evasion, Infectivity, and Fusogenicity of SARS-CoV-2 Omicron BA.2.86 and FLip Variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Tsujino, S.; Deguchi, S.; Nomai, T.; Padilla-Blanco, M.; Plianchaisuk, A.; Wang, L.; Begum, M.M.; Uriu, K.; Mizuma, K.; Nao, N.; et al. Virological Characteristics of the SARS-CoV-2 Omicron EG.5.1 Variant. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gili, R.; Burioni, R. SARS-CoV-2 before and after Omicron: Two Different Viruses and Two Different Diseases? J. Transl. Med. 2023, 21, 251. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Infectivity and Fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2 Spike. Cell 2022, 185, 2103–2115.e19. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron Is an Immune Escape Variant with an Altered Cell Entry Pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Pommerenke, C.; Rand, U.; Uphoff, C.C.; Nagel, S.; Zaborski, M.; Hauer, V.; Kaufmann, M.; Meyer, C.; Denkmann, S.A.; Riese, P.; et al. Identification of Cell Lines CL-14, CL-40 and CAL-51 as Suitable Models for SARS-CoV-2 Infection Studies. PLoS ONE 2021, 16, e0255622. [Google Scholar] [CrossRef]
- Xia, H.; Yeung, J.; Kalveram, B.; Bills, C.J.; Chen, J.Y.-C.; Kurhade, C.; Zou, J.; Widen, S.G.; Mann, B.R.; Kondor, R.; et al. Cross-Neutralization and Viral Fitness of SARS-CoV-2 Omicron Sublineages. Emerg. Microbes Infect. 2023, 12, e2161422. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, M.; Wei, F.; Huang, S.; Xu, J. COVID’s Future: Viral Multi-Lineage Evolution and the Dynamics of Small Epidemic Waves without Seasonality in COVID-19. J. Biosaf. Biosecurity 2023, 5, 96–99. [Google Scholar] [CrossRef]
- Troyano-Hernáez, P.; Reinosa, R.; Holguín, Á. Evolution of SARS-CoV-2 Envelope, Membrane, Nucleocapsid, and Spike Structural Proteins from the Beginning of the Pandemic to September 2020: A Global and Regional Approach by Epidemiological Week. Viruses 2021, 13, 243. [Google Scholar] [CrossRef]
- Tarig, M.S.A.; Ullah, M.F.; Elssaig, E.H.; Ahmed-Abakur, E.H. Unique SARS-CoV-2 Variant Exhibiting Plenteous Missense Mutations in Structural and Nonstructural Genes. Cytol. Genet. 2021, 55, 606–612. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-Wide Analysis of SARS-CoV-2 Virus Strains Circulating Worldwide Implicates Heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.-Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid Assessment of SARS-CoV-2-Evolved Variants Using Virus-like Particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid Mutations in SARS-CoV-2 Augment Replication and Pathogenesis. PLoS Pathog. 2022, 18, e1010627. [Google Scholar] [CrossRef]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 Envelope and Membrane Proteins: Structural Differences Linked to Virus Characteristics? BioMed Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef]
- Xia, B.; Wang, Y.; Pan, X.; Cheng, X.; Ji, H.; Zuo, X.; Jiang, H.; Li, J.; Gao, Z. Why Is the SARS-CoV-2 Omicron Variant Milder? Innovation 2022, 3, 100251. [Google Scholar] [CrossRef]
- Cruz, C.A.K.; Medina, P.M.B. Temporal Changes in the Accessory Protein Mutations of SARS-CoV-2 Variants and Their Predicted Structural and Functional Effects. J. Med. Virol. 2022, 94, 5189–5200. [Google Scholar] [CrossRef]
- Liu, X.; Guo, L.; Xu, T.; Lu, X.; Ma, M.; Sheng, W.; Wu, Y.; Peng, H.; Cao, L.; Zheng, F.; et al. A Comprehensive Evolutionary and Epidemiological Characterization of Insertion and Deletion Mutations in SARS-CoV-2 Genomes. Virus Evol. 2021, 7, veab104. [Google Scholar] [CrossRef]
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and Functions of SARS-CoV-2 Proteins in Host Immune Evasion. Front. Immunol. 2022, 13, 940756. [Google Scholar] [CrossRef]
- McGrath, M.E.; Xue, Y.; Dillen, C.; Oldfield, L.; Assad-Garcia, N.; Zaveri, J.; Singh, N.; Baracco, L.; Taylor, L.J.; Vashee, S.; et al. SARS-CoV-2 Variant Spike and Accessory Gene Mutations Alter Pathogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2204717119. [Google Scholar] [CrossRef]
- Taha, T.Y.; Chen, I.P.; Hayashi, J.M.; Tabata, T.; Walcott, K.; Kimmerly, G.R.; Syed, A.M.; Ciling, A.; Suryawanshi, R.K.; Martin, H.S.; et al. Rapid Assembly of SARS-CoV-2 Genomes Reveals Attenuation of the Omicron BA.1 Variant through NSP6. Nat. Commun. 2023, 14, 2308. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Pang, X.; Li, P.; Zhang, L.; Que, L.; Dong, M.; Xie, B.; Wang, Q.; Wei, Y.; Xie, X.; Li, L.; et al. Emerging Severe Acute Respiratory Syndrome Coronavirus 2 Mutation Hotspots Associated with Clinical Outcomes and Transmission. Front. Microbiol. 2021, 12, 753823. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Y.; Chin, C.V.; Kenney, D.; Tavares, A.H.; Khan, N.; Conway, H.L.; Liu, G.; Choudhary, M.C.; Gertje, H.P.; O’Connell, A.K.; et al. Spike and Nsp6 Are Key Determinants of SARS-CoV-2 Omicron BA.1 Attenuation. Nature 2023, 615, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Selvaraj, P.; Sangare, K.; Luan, B.; Wang, T.T. Spike Protein-Independent Attenuation of SARS-CoV-2 Omicron Variant in Laboratory Mice. Cell Rep. 2022, 40, 111359. [Google Scholar] [CrossRef]
- Zhou, Z.; Huang, C.; Zhou, Z.; Huang, Z.; Su, L.; Kang, S.; Chen, X.; Chen, Q.; He, S.; Rong, X.; et al. Structural Insight Reveals SARS-CoV-2 ORF7a as an Immunomodulating Factor for Human CD14+ Monocytes. iScience 2021, 24, 102187. [Google Scholar] [CrossRef]
- García-García, T.; Fernández-Rodríguez, R.; Redondo, N.; de Lucas-Rius, A.; Zaldívar-López, S.; López-Ayllón, B.D.; Suárez-Cárdenas, J.M.; Jiménez-Marín, Á.; Montoya, M.; Garrido, J.J. Impairment of Antiviral Immune Response and Disruption of Cellular Functions by SARS-CoV-2 ORF7a and ORF7b. iScience 2022, 25, 105444. [Google Scholar] [CrossRef]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.; Low, J.G.H.; Tan, C.W.; et al. Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2. mBio 2020, 11, e01610–e01620. [Google Scholar] [CrossRef]
- Mishra, S. Computational Structural and Functional Analyses of ORF10 in Novel Coronavirus SARS-CoV-2 Variants to Understand Evolutionary Dynamics. Evol. Bioinforma. Online 2022, 18, 11769343221108218. [Google Scholar] [CrossRef]
- Shehata, A.A.; Attia, Y.A.; Rahman, M.T.; Basiouni, S.; El-Seedi, H.R.; Azhar, E.I.; Khafaga, A.F.; Hafez, H.M. Diversity of Coronaviruses with Particular Attention to the Interspecies Transmission of SARS-CoV-2. Animals 2022, 12, 378. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, D.; Han, P.; Kang, X.; Zheng, A.; Xu, Z.; Zhao, X.; Wang, V.Y.-F.; Qi, J.; Wang, Q.; et al. Structural Basis of SARS-CoV-2 and Its Variants Binding to Intermediate Horseshoe Bat ACE2. Int. J. Biol. Sci. 2022, 18, 4658–4668. [Google Scholar] [CrossRef]
- Gao, G.F.; Wang, L. COVID-19 Expands Its Territories from Humans to Animals. China CDC Wkly. 2021, 3, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Allender, M.C.; Adkesson, M.J.; Langan, J.N.; Delk, K.W.; Meehan, T.; Aitken-Palmer, C.; McEntire, M.M.; Killian, M.L.; Torchetti, M.; Morales, S.A.; et al. Multi-species Outbreak of SARS-CoV-2 Delta Variant in a Zoological Institution, with the Detection in Two New Families of Carnivores. Transbound. Emerg. Dis. 2022, 69, E3060–E3075. [Google Scholar] [CrossRef]
- Du, P.; Gao, G.F.; Wang, Q. The Mysterious Origins of the Omicron Variant of SARS-CoV-2. Innovation 2022, 3, 100206. [Google Scholar] [CrossRef]
- Carlson, C.J.; Albery, G.F.; Merow, C.; Trisos, C.H.; Zipfel, C.M.; Eskew, E.A.; Olival, K.J.; Ross, N.; Bansal, S. Climate Change Increases Cross-Species Viral Transmission Risk. Nature 2022, 607, 555–562. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, X.; Zhang, W.; Wang, X.; Zhang, D.; Wang, J.; Liu, H.; Ren, Z.; Ruktanonchai, N.W.; Ruktanonchai, C.W.; et al. Effects of Public-Health Measures for Zeroing out Different SARS-CoV-2 Variants. Nat. Commun. 2023, 14, 5270. [Google Scholar] [CrossRef]
- Wong, C.K.H.; Au, I.C.H.; Lau, K.T.K.; Lau, E.H.Y.; Cowling, B.J.; Leung, G.M. Real-World Effectiveness of Early Molnupiravir or Nirmatrelvir-Ritonavir in Hospitalised Patients with COVID-19 without Supplemental Oxygen Requirement on Admission during Hong Kong’s Omicron BA.2 Wave: A Retrospective Cohort Study. Lancet Infect. Dis. 2022, 22, 1681–1693. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.-C.; et al. Multiple Pathways for SARS-CoV-2 Resistance to Nirmatrelvir. Nature 2023, 613, 558–564. [Google Scholar] [CrossRef]
- Boby, M.L.; Fearon, D.; Ferla, M.; Filep, M.; Koekemoer, L.; Robinson, M.C.; COVID Moonshot Consortium‡; Chodera, J.D.; Lee, A.A.; London, N.; et al. Open Science Discovery of Potent Noncovalent SARS-CoV-2 Main Protease Inhibitors. Science 2023, 382, eabo7201. [Google Scholar] [CrossRef]
- Liu, J.; Mao, F.; Chen, J.; Lu, S.; Qi, Y.; Sun, Y.; Fang, L.; Yeung, M.L.; Liu, C.; Yu, G.; et al. An IgM-like Inhalable ACE2 Fusion Protein Broadly Neutralizes SARS-CoV-2 Variants. Nat. Commun. 2023, 14, 5191. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Huang, Z.; Xiao, J.; Wu, Y.; Xia, N.; Yuan, Q. Evolution of the SARS-CoV-2 Omicron Variants: Genetic Impact on Viral Fitness. Viruses 2024, 16, 184. https://doi.org/10.3390/v16020184
Liu W, Huang Z, Xiao J, Wu Y, Xia N, Yuan Q. Evolution of the SARS-CoV-2 Omicron Variants: Genetic Impact on Viral Fitness. Viruses. 2024; 16(2):184. https://doi.org/10.3390/v16020184
Chicago/Turabian StyleLiu, Wenhao, Zehong Huang, Jin Xiao, Yangtao Wu, Ningshao Xia, and Quan Yuan. 2024. "Evolution of the SARS-CoV-2 Omicron Variants: Genetic Impact on Viral Fitness" Viruses 16, no. 2: 184. https://doi.org/10.3390/v16020184
APA StyleLiu, W., Huang, Z., Xiao, J., Wu, Y., Xia, N., & Yuan, Q. (2024). Evolution of the SARS-CoV-2 Omicron Variants: Genetic Impact on Viral Fitness. Viruses, 16(2), 184. https://doi.org/10.3390/v16020184