
Friends and Foes: The Ambivalent Role of Autophagy in HIV-1 Infection


Abstract
:1. Introduction
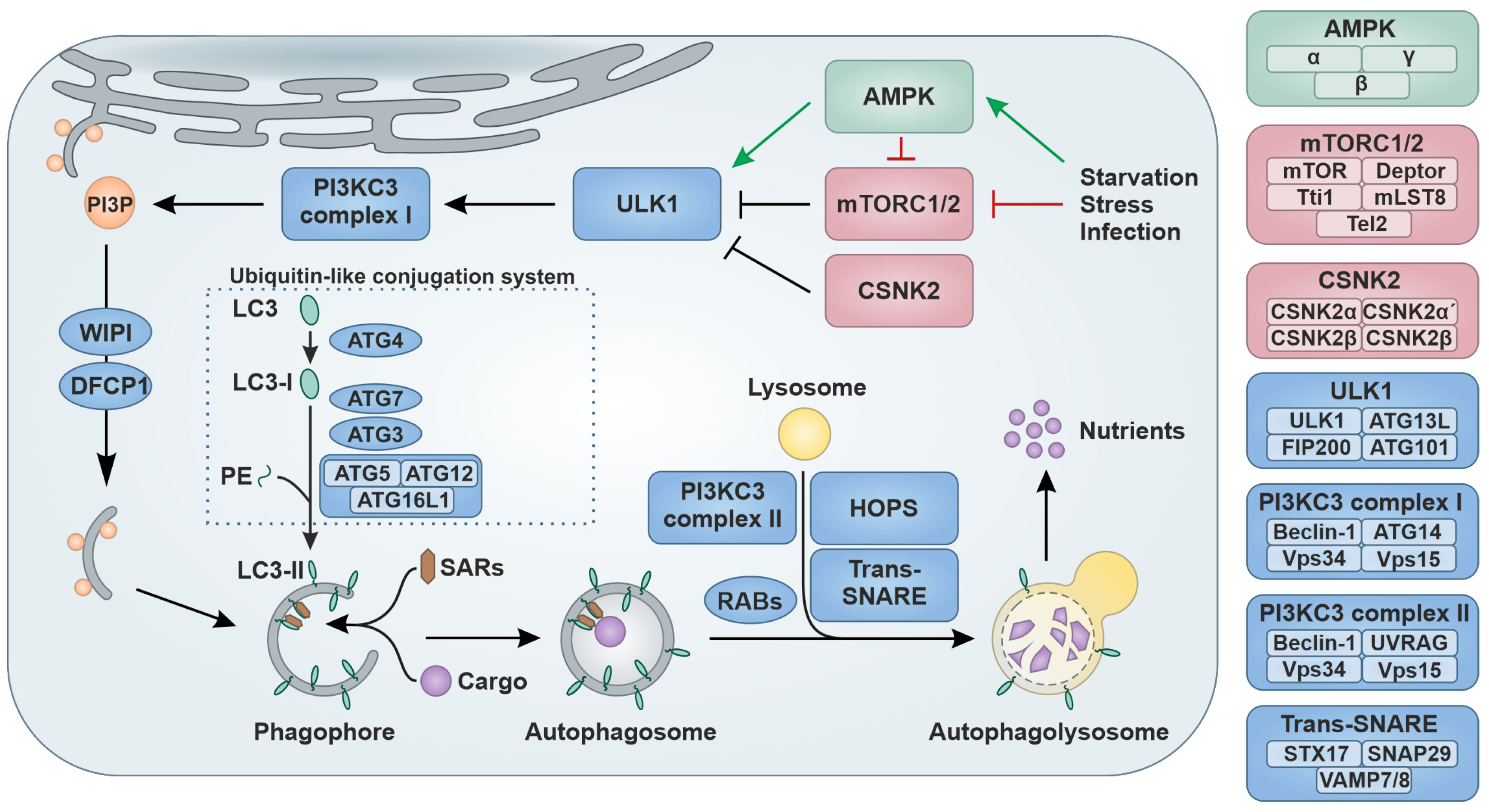
1.1. Regulation of Autophagy
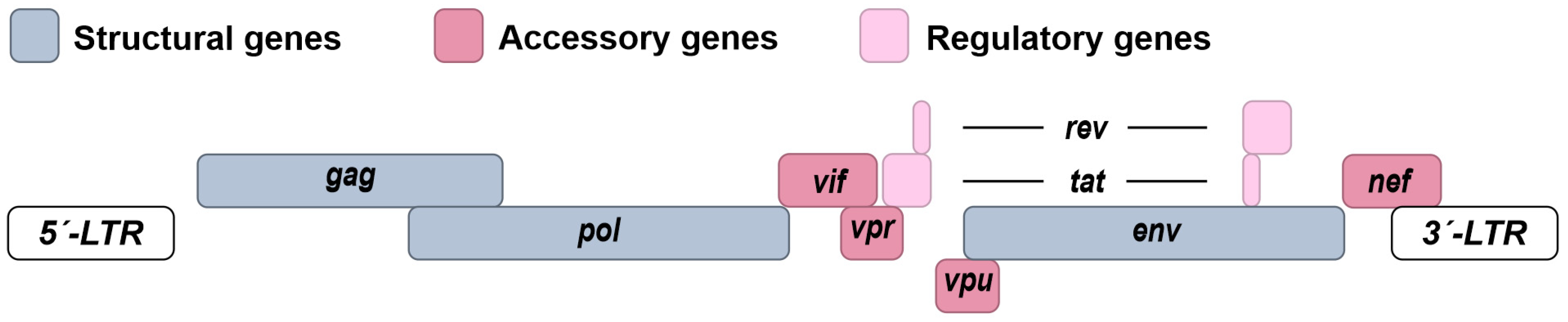
1.2. Molecular Biology of HIV-1
2. The Interplay between Autophagy and HIV-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Protein | Targeted by Autophagy | Impact on Autophagy |
|---|---|---|
| Gag | HDAC6-mediated autophagic degradation of p55 in transfected HEK293T [83,84]. Degradation of p24 by autophagy in a TRIM5α-dependent manner in rhesus CD4+ T cells and Langerhans cells [20,118]. | |
| Vif | HDAC6-mediated autophagic degradation in transfected HEK293T [82,84]. |
|
| Vpr | ||
| Tat | Degradation in a p62-dependent manner in CD4+ T cells, potentially ubiquitin dependent [85,86]. |
|
| Vpu |
| |
| Env |
| |
| Nef |
| |
| ASP |
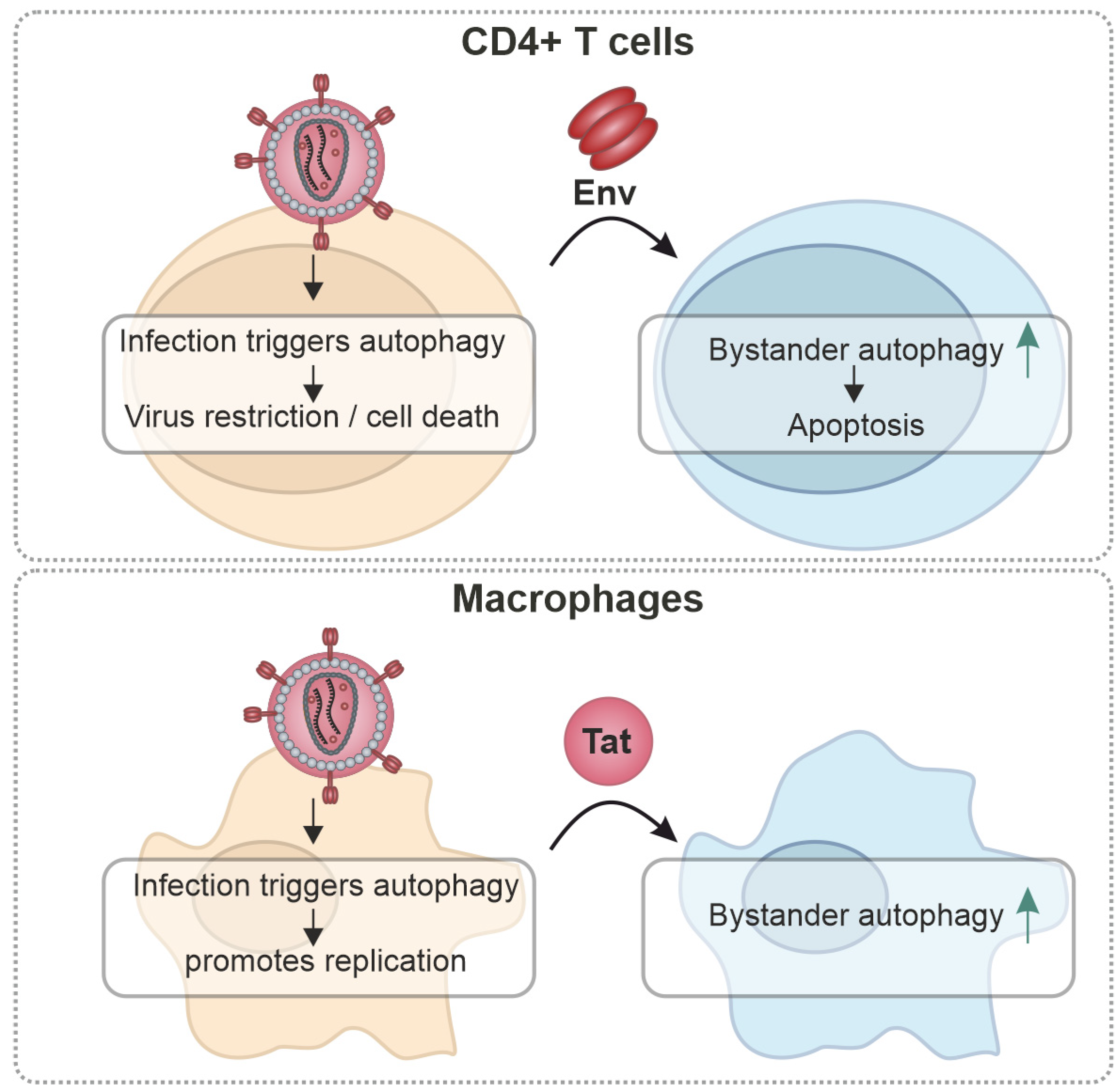
3. Cell-Type-Specific Effects
4. Autophagy Modulation as an Antiviral Approach
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less than a Decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular Machinery for Self-Eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Roach, P.J. AMPK → ULK1 → Autophagy. Mol. Cell Biol. 2011, 31, 3082–3084. [Google Scholar] [CrossRef] [PubMed]
- Hoenigsperger, H.; Koepke, L.; Acharya, D.; Hunszinger, V.; Freisem, D.; Grenzner, A.; Wiese, S.; Kirchhoff, F.; Gack, M.U.; Sparrer, K.M.J. CSNK2 Suppresses Autophagy by Activating FLN-NHL-Containing TRIM Proteins. Autophagy 2023. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Munson, M.J.; Ganley, I.G. MTOR, PIK3C3, and Autophagy: Signaling the Beginning from the End. Autophagy 2015, 11, 2375–2376. [Google Scholar] [CrossRef] [PubMed]
- Nähse, V.; Raiborg, C.; Tan, K.W.; Mørk, S.; Torgersen, M.L.; Wenzel, E.M.; Nager, M.; Salo, V.T.; Johansen, T.; Ikonen, E.; et al. ATPase Activity of DFCP1 Controls Selective Autophagy. Nat. Commun. 2023, 14, 4051. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Marafi, D.; El-Hattab, A.W. WIPI Proteins: Biological Functions and Related Syndromes. Front. Mol. Neurosci. 2022, 15, 1011918. [Google Scholar] [CrossRef] [PubMed]
- Mailler, E.; Guardia, C.M.; Bai, X.; Jarnik, M.; Williamson, C.D.; Li, Y.; Maio, N.; Golden, A.; Bonifacino, J.S. The Autophagy Protein ATG9A Enables Lipid Mobilization from Lipid Droplets. Nat. Commun. 2021, 12, 6750. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Klionsky, D.J. Autophagic Membrane Delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, D.S.; Otto, N.M.; Park, J.-M.; Song, D.; Kim, D.-H. GABARAPs and LC3s Have Opposite Roles in Regulating ULK1 for Autophagy Induction. Autophagy 2020, 16, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An Autophagy-Related Ubiquitin-like Protein Family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Gubas, A.; Dikic, I. A Guide to the Regulation of Selective Autophagy Receptors. FEBS J. 2022, 289, 75–89. [Google Scholar] [CrossRef]
- Kimura, T.; Mandell, M.; Deretic, V. Precision Autophagy Directed by Receptor Regulators—Emerging Examples within the TRIM Family. J. Cell Sci. 2016, 129, 881–891. [Google Scholar] [CrossRef]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM Proteins Regulate Autophagy and Can Target Autophagic Substrates by Direct Recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.J.; Gack, M.U. TRIM Proteins: New Players in Virus-Induced Autophagy. PLoS Pathog. 2018, 14, e1006787. [Google Scholar] [CrossRef]
- Koepke, L.; Gack, M.U.; Sparrer, K.M. The Antiviral Activities of TRIM Proteins. Curr. Opin. Microbiol. 2021, 59, 50–57. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Hou, C.; Lai, Y.; Long, J.; Liu, J.; Zhong, Q.; Diao, J. SNARE-Mediated Membrane Fusion in Autophagy. Semin. Cell Dev. Biol. 2016, 60, 97–104. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of Autophagy by the Rab GTPase Network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The Role of Autophagy in Neurodegenerative Disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Yang, Y.; Klionsky, D.J. Autophagy and Disease: Unanswered Questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and Cancer Therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during Viral Infection—A Double-Edged Sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Levine, B. Viruses and Autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef]
- Mao, K.; Klionsky, D.J. Xenophagy: A Battlefield between Host and Microbe, and a Possible Avenue for Cancer Treatment. Autophagy 2016, 13, 223–224. [Google Scholar] [CrossRef]
- Thurston, T.L.M.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 Adaptor and Autophagy Receptor NDP52 Restricts the Proliferation of Ubiquitin-Coated Bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The Adaptor Protein P62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef]
- Jassey, A.; Jackson, W.T. Viruses and Autophagy: Bend, but Don’t Break. Nat. Rev. Microbiol. 2023, 1–13. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like Receptors Control Autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Cadwell, K. Crosstalk between Autophagy and Inflammatory Signalling Pathways: Balancing Defence and Homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and Its Role in MHC-Mediated Antigen Presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Koepke, L.; Hirschenberger, M.; Hayn, M.; Kirchhoff, F.; Sparrer, K.M. Manipulation of Autophagy by SARS-CoV-2 Proteins. Autophagy 2021, 17, 2659–2661. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic Functional Analysis of SARS-CoV-2 Proteins Uncovers Viral Innate Immune Antagonists and Remaining Vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef] [PubMed]
- Killian, M.S. Dual Role of Autophagy in HIV-1 Replication and Pathogenesis. AIDS Res. Ther. 2012, 9, 16. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Gallo, R.C.; Sarin, P.S.; Gelmann, E.P.; Robert-Guroff, M.; Richardson, E.; Kalyanaraman, V.S.; Mann, D.; Sidhu, G.D.; Stahl, R.E.; Zolla-Pazner, S.; et al. Isolation of Human T-Cell Leukemia Virus in Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- HIV and AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 15 February 2024).
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. Molecular Mechanisms of HIV Entry. Adv. Exp. Med. Biol. 2012, 726, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 Latency Provides Clues for the Eradication of Long-Term Reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Ruelas, D.S.; Greene, W.C. An Integrated Overview of HIV-1 Latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Siliciano, R.F. Nuclear Landscape of HIV-1 Infection and Integration. Nat. Rev. Microbiol. 2017, 15, 69–82. [Google Scholar] [CrossRef]
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. SnapShot: Antiviral Restriction Factors. Cell 2015, 163, 774–774.e1. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, S.; Xu, M.; He, Y.; Zhang, X.; Xiong, Y.; Sun, H.; Ding, H.; Geng, W.; Shang, H.; et al. Vpr Counteracts the Restriction of LAPTM5 to Promote HIV-1 Infection in Macrophages. Nat. Commun. 2021, 12, 3691. [Google Scholar] [CrossRef]
- Laliberté, A.; Prelli Bozzo, C.; Stahl-Hennig, C.; Hunszinger, V.; Joas, S.; Sauermann, U.; Roshani, B.; Klippert, A.; Daskalaki, M.; Mätz-Rensing, K.; et al. Vpr Attenuates Antiviral Immune Responses and Is Critical for Full Pathogenicity of SIVmac239 in Rhesus Macaques. iScience 2023, 26, 108351. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Alves, C. HIV-1 Nef Targets Restriction Factors. Nat. Rev. Microbiol. 2015, 13, 661. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Volcic, M.; Wiesmüller, L.; Kirchhoff, F. Small but Highly Versatile: The Viral Accessory Protein Vpu. Annu. Rev. Virol. 2023, 10, 243–259. [Google Scholar] [CrossRef]
- Volcic, M.; Sparrer, K.M.J.; Koepke, L.; Hotter, D.; Sauter, D.; Stürzel, C.M.; Scherer, M.; Stamminger, T.; Hofmann, T.G.; Arhel, N.J.; et al. Vpu Modulates DNA Repair to Suppress Innate Sensing and Hyper-Integration of HIV-1. Nat. Microbiol. 2020, 5, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Wildum, S.; Schindler, M.; Münch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 Down-Modulation and Resistance of Human Immunodeficiency Virus Type 1-Infected T Cells to Superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 Replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef]
- Nchioua, R.; Bosso, M.; Kmiec, D.; Kirchhoff, F. Cellular Factors Targeting HIV-1 Transcription and Viral RNA Transcripts. Viruses 2020, 12, 495. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retroviruses 2018, 34, 739–759. [Google Scholar] [CrossRef]
- Pennings, P.S. HIV Drug Resistance: Problems and Perspectives. Infect. Dis. Rep. 2013, 5, e5. [Google Scholar] [CrossRef] [PubMed]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Lingappa, J.R.; Lingappa, V.R.; Reed, J.C. Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses 2021, 13, 451. [Google Scholar] [CrossRef]
- Roche, M.; Salimi, H.; Duncan, R.; Wilkinson, B.L.; Chikere, K.; Moore, M.S.; Webb, N.E.; Zappi, H.; Sterjovski, J.; Flynn, J.K.; et al. A Common Mechanism of Clinical HIV-1 Resistance to the CCR5 Antagonist Maraviroc despite Divergent Resistance Levels and Lack of Common Gp120 Resistance Mutations. Retrovirology 2013, 10, 43. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV Infection. Nat. Rev. Dis. Primers 2015, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV: Shock and Kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “Shock and Kill” Therapy: In Need of Revision. Antiviral Res. 2019, 166, 19–34. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Tan, J.; Devadas, K.; Ragupathy, V.; Takeda, K.; Zhao, J.; Hewlett, I. HIV-1 and HIV-2 Infections Induce Autophagy in Jurkat and CD4+ T Cells. Cell. Signal. 2012, 24, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Viral Evasion of Autophagy. Autophagy 2008, 4, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.P.; De Simone, F.I.; Iorio, V.; De Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. HIV-1 Tat Protein Induces Glial Cell Autophagy through Enhancement of BAG3 Protein Levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef]
- Wu, X.; Dong, H.; Ye, X.; Zhong, L.; Cao, T.; Xu, Q.; Wang, J.; Zhang, Y.; Xu, J.; Wang, W.; et al. HIV-1 Tat Increases BAG3 via NF-κB Signaling to Induce Autophagy during HIV-Associated Neurocognitive Disorder. Cell Cycle 2018, 17, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Niu, F.; Hu, G.; Guo, M.-L.; Sil, S.; Buch, S. HIV Tat-Mediated Induction of Autophagy Regulates the Disruption of ZO-1 in Brain Endothelial Cells. Tissue Barriers 2020, 8, 1748983. [Google Scholar] [CrossRef]
- Yang, G.; Li, J.; Leung, C.-K.; Shen, B.; Wang, C.; Xu, Y.; Lin, S.; Zhang, S.; Tan, Y.; Zhang, H.; et al. Methamphetamine and HIV-1 Tat Proteins Synergistically Induce Microglial Autophagy via Activation of the Nrf2/NQO1/HO-1 Signal Pathway. Neuropharmacology 2022, 220, 109256. [Google Scholar] [CrossRef]
- Zhou, H.; Zheng, Y.; He, Y.; Chen, Z.; He, B. The Role of Autophagy in THP-1 Macrophages Resistance to HIV- Vpr-Induced Apoptosis. Exp. Cell Res. 2017, 351, 68–73. [Google Scholar] [CrossRef]
- Mazzuca, P.; Marsico, S.; Schulze, K.; Mitola, S.; Pils, M.C.; Giagulli, C.; Guzman, C.A.; Caruso, A.; Caccuri, F. Role of Autophagy in HIV-1 Matrix Protein P17-Driven Lymphangiogenesis. J. Virol. 2017, 91, e00801-17. [Google Scholar] [CrossRef]
- Valera, M.-S.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; García-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G Complex Regulates HIV-1 Infectiveness by Inducing Vif Autophagic Degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef]
- Marrero-Hernández, S.; Márquez-Arce, D.; Cabrera-Rodríguez, R.; Estévez-Herrera, J.; Pérez-Yanes, S.; Barroso-González, J.; Madrid, R.; Machado, J.-D.; Blanco, J.; Valenzuela-Fernández, A. HIV-1 Nef Targets HDAC6 to Assure Viral Production and Virus Infection. Front. Microbiol. 2019, 10, 2437. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Lorenzo-Sánchez, I.; Estévez-Herrera, J.; García-Luis, J.; Trujillo-González, R.; Valenzuela-Fernández, A. TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. Int. J. Mol. Sci. 2023, 24, 7658. [Google Scholar] [CrossRef]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy Restricts HIV-1 Infection by Selectively Degrading Tat in CD4 + T Lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates, J.R.; Jones, K.A. SHMT2 and the BRCC36/BRISC Deubiquitinase Regulate HIV-1 Tat K63-Ubiquitylation and Destruction by Autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Müller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy Promotes MHC Class II Presentation of Peptides from Intracellular Source Proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef]
- Delgado, M.; Singh, S.; De Haro, S.; Master, S.; Ponpuak, M.; Dinkins, C.; Ornatowski, W.; Vergne, I.; Deretic, V. Autophagy and Pattern Recognition Receptors in Innate Immunity. Immunol. Rev. 2009, 227, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Münz, C. Innate and Adaptive Immunity through Autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of Autophagy in HIV Infection and Pathogenesis. J. Intern. Med. 2017, 281, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef Counteracts Autophagy Restriction by Enhancing the Association between BECN1 and Its Inhibitor BCL2 in a PRKN-Dependent Manner. Autophagy 2021, 17, 553–577. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Simpson, S.; Shi, Y.; Chen, Y.; Benjamin, J.; Serra-Moreno, R. HIV Nef-Mediated Ubiquitination of BCL2: Implications in Autophagy and Apoptosis. Front. Immunol. 2021, 12, 682624. [Google Scholar] [CrossRef]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Young, L.N.; Morris, K.L.; von Bülow, S.; Schöneberg, J.; Yamamoto-Imoto, H.; Oe, Y.; Yamamoto, K.; Nakamura, S.; Stjepanovic, G.; et al. Bidirectional Control of Autophagy by BECN1 BARA Domain Dynamics. Mol. Cell 2019, 73, 339–353.e6. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Teng, J.; Chen, J. New Insights Regarding SNARE Proteins in Autophagosome-Lysosome Fusion. Autophagy 2021, 17, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jain, A.; Farzam, F.; Jia, J.; Gu, Y.; Choi, S.W.; Mudd, M.H.; Claude-Taupin, A.; Wester, M.J.; Lidke, K.A.; et al. Mechanism of Stx17 Recruitment to Autophagosomes via IRGM and Mammalian Atg8 Proteins. J. Cell Biol. 2018, 217, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM Is a Common Target of RNA Viruses That Subvert the Autophagy Network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Alfaisal, J.; Machado, A.; Galais, M.; Robert-Hebmann, V.; Arnauné-Pelloquin, L.; Espert, L.; Biard-Piechaczyk, M. HIV-1 Vpr Inhibits Autophagy during the Early Steps of Infection of CD4 T Cells. Biol. Cell 2019, 111, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, Y.; Wen, X.; Yin, Z.; Zhang, Z.; Klionsky, D.J. How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems. Antioxidants 2022, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Huang, Q.-Q.; Birkett, R.; Doyle, R.; Dorfleutner, A.; Stehlik, C.; He, C.; Pope, R.M. SNAPIN Is Critical for Lysosomal Acidification and Autophagosome Maturation in Macrophages. Autophagy 2017, 13, 285–301. [Google Scholar] [CrossRef]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Callen, S.; Buch, S.; Sawaya, B.E. HIV-1 Vpr Protein Impairs Lysosome Clearance Causing SNCA/Alpha-Synuclein Accumulation in Neurons. Autophagy 2021, 17, 1768–1782. [Google Scholar] [CrossRef]
- Borel, S.; Robert-Hebmann, V.; Alfaisal, J.; Jain, A.; Faure, M.; Espert, L.; Chaloin, L.; Paillart, J.-C.; Johansen, T.; Biard-Piechaczyk, M. HIV-1 Viral Infectivity Factor Interacts with Microtubule-Associated Protein Light Chain 3 and Inhibits Autophagy. AIDS 2015, 29, 275–286. [Google Scholar] [CrossRef]
- Madjo, U.; Leymarie, O.; Frémont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. LC3C Contributes to Vpu-Mediated Antagonism of BST2/Tetherin Restriction on HIV-1 Release through a Non-Canonical Autophagy Pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef]
- Liu, Z.; Torresilla, C.; Xiao, Y.; Nguyen, P.T.; Caté, C.; Barbosa, K.; Rassart, É.; Cen, S.; Bourgault, S.; Barbeau, B. HIV-1 Antisense Protein of Different Clades Induces Autophagy and Associates with the Autophagy Factor P62. J. Virol. 2019, 93, e01757-18. [Google Scholar] [CrossRef]
- Torresilla, C.; Larocque, É.; Landry, S.; Halin, M.; Coulombe, Y.; Masson, J.-Y.; Mesnard, J.-M.; Barbeau, B. Detection of the HIV-1 Minus-Strand-Encoded Antisense Protein and Its Association with Autophagy. J. Virol. 2013, 87, 5089–5105. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Au, K.; Fang, J.; Yim, H.C.; Chow, K.; Ho, P.; Lau, A.S. HIV-1 Trans-Activator Protein Dysregulates IFN-γ Signaling and Contributes to the Suppression of Autophagy Induction. AIDS 2011, 25, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Dumaop, W.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat Alters Neuronal Autophagy by Modulating Autophagosome Fusion to the Lysosome: Implications for HIV-Associated Neurocognitive Disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-S.; Zhang, Z.-G.; Zhou, Z.; Du, G.-Y.; Li, H.; Yu, X.-Y.; Huang, Y.-H. PKM2-Mediated Inhibition of Autophagy Facilitates Tat’s Inducing HIV-1 Transactivation. Arch. Biochem. Biophys. 2017, 625–626, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Buonaguro, L.; Barillari, G.; Fiorelli, V.; Gendelman, R.; Morgan, R.A.; Wingfield, P.; Gallo, R.C. Release, Uptake, and Effects of Extracellular Human Immunodeficiency Virus Type 1 Tat Protein on Cell Growth and Viral Transactivation. J. Virol. 1993, 67, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Robinson, S.M.; Nath, A. Permeability of the Blood–Brain Barrier to HIV-1 Tat. Exp. Neurol. 2005, 193, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A.; He, J.J. Uptake of HIV-1 Tat Protein Mediated by Low-Density Lipoprotein Receptor-Related Protein Disrupts the Neuronal Metabolic Balance of the Receptor Ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef]
- Hui, L.; Chen, X.; Haughey, N.J.; Geiger, J.D. Role of Endolysosomes in HIV-1 Tat-Induced Neurotoxicity. ASN Neuro 2012, 4, AN20120017. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-Mediated Microglial Activation: Role of Mitochondrial Dysfunction and Defective Mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef] [PubMed]
- Omeragic, A.; Kayode, O.; Hoque, M.T.; Bendayan, R. Potential Pharmacological Approaches for the Treatment of HIV-1 Associated Neurocognitive Disorders. Fluids Barriers CNS 2020, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy Pathway Intersects with HIV-1 Biosynthesis and Regulates Viral Yields in Macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy Is Involved in T Cell Death after Binding of HIV-1 Envelope Proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human Immunodeficiency Virus-1 Inhibition of Immunoamphisomes in Dendritic Cells Impairs Early Innate and Adaptive Immune Responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; Van Hamme, J.L.; Tigchelaar, W.; Van Der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor Usage Dictates HIV-1 Restriction by Human TRIM5α in Dendritic Cell Subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 Inhibits Autophagy in Bystander Macrophage/Monocytic Cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential Role of Autophagy in CD4 T Cells and Macrophages during X4 and R5 HIV-1 Infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy Is a Critical Regulator of Memory CD8(+) T Cell Formation. eLife 2014, 3, e03706. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.-H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy Is Essential for Effector CD8(+) T Cell Survival and Memory Formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef]
- Botbol, Y.; Guerrero-Ros, I.; Macian, F. Key Roles of Autophagy in the Regulation of T-Cell Function. Eur. J. Immunol. 2016, 46, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Murera, D.; Arbogast, F.; Arnold, J.; Bouis, D.; Muller, S.; Gros, F. CD4 T Cell Autophagy Is Integral to Memory Maintenance. Sci. Rep. 2018, 8, 5951. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.-U. Regulation of the Innate Immune System by Autophagy: Monocytes, Macrophages, Dendritic Cells and Antigen Presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Lu, J.-H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef]
- Spector, S.A.; Zhou, D. Autophagy: An Overlooked Mechanism of HIV-1 Pathogenesis and neuroAIDS? Autophagy 2008, 4, 704–706. [Google Scholar] [CrossRef]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Elisabet Garcia, E.G.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 Gp41 Fusogenic Function Triggers Autophagy in Uninfected Cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of Autophagy in HIV Infection. Immunol. Cell Biol. 2015, 93, 11–17. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Law, B.K. Rapamycin: An Anti-Cancer Immunosuppressant? Crit. Rev. Oncol. Hematol. 2005, 56, 47–60. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-Yl)-3-(Trifluoromethyl)Phenyl)-9-(Quinolin-3-Yl)Benzo[h][1,6]Naphthyridin-2(1H)-One as a Highly Potent, Selective Mammalian Target of Rapamycin (mTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Rapalogs Potential as Practical Alternatives to Rapamycin. ACS Med. Chem. Lett. 2019, 10, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-M.; Lin, P.-M.; Tsai, Y.-T.; Tsai, M.-S.; Tseng, C.-H.; Lin, S.-F.; Yang, M.-Y. NVP-BEZ235, a Dual PI3K-mTOR Inhibitor, Suppresses the Growth of FaDu Hypopharyngeal Squamous Cell Carcinoma and Has a Synergistic Effect with Cisplatin. Cell Death Discov. 2018, 4, 1–10. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.-S.; Li, X.-O.; Du, S.-Y. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. Autophagy Inhibitors. Cell Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 Inhibitor Blocks Autophagy and Uncovers a Role for NCOA4 in Ferritin Degradation and Iron Homeostasis in Vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of P53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells 2021, 10, 1798. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a Candidate Therapeutic Autophagy–Inducing Peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Pedreño-López, S.; García, E.; Guerrero, D.; Gómez-Mora, E.; Molina Mateu, L.; Orera Pérez, F.; Senserrich, J.; Clotet, B.; Cabrera, C. Modulation of the Autophagic Pathway Inhibits HIV-1 Infection in Human Lymphoid Tissue Cultured Ex Vivo. Sci. Rep. 2022, 12, 7439. [Google Scholar] [CrossRef]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.-L.; Spector, S.A. Autophagy Induction by Histone Deacetylase Inhibitors Inhibits HIV Type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef]
- Campbell, G.R.; Bruckman, R.S.; Herns, S.D.; Joshi, S.; Durden, D.L.; Spector, S.A. Induction of Autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 Inhibitors Suppresses HIV-1 Replication. J. Biol. Chem. 2018, 293, 5808–5820. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Hon, S.; Teodorof-Diedrich, C.; Spector, S.A. Trehalose Inhibits Human Immunodeficiency Virus Type 1 Infection in Primary Human Macrophages and CD4+ T Lymphocytes through Two Distinct Mechanisms. J. Virol. 2020, 94, e00237-20. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; To, R.K.; Zhang, G.; Spector, S.A. SMAC Mimetics Induce Autophagy-Dependent Apoptosis of HIV-1-Infected Macrophages. Cell Death Dis. 2020, 11, 590. [Google Scholar] [CrossRef]
- Imamichi, T.; Goswami, S.; Hu, X.; Laverdure, S.; Yang, J.; Qiu, J.; Chen, Q.; Sherman, B.T.; Chang, W. MicroRNA Profiles in Monocyte-Derived Macrophages Generated by Interleukin-27 and Human Serum: Identification of a Novel HIV-Inhibiting and Autophagy-Inducing MicroRNA. Int. J. Mol. Sci. 2021, 22, 1290. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Ma, Z.; Zhang, M.; Rushdi, M.N.; Krueger, C.J.; Chen, A.K. Inhibition of Retroviral Gag Assembly by Non-Silencing miRNAs Promotes Autophagic Viral Degradation. Protein Cell 2018, 9, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef] [PubMed]
- Cloherty, A.P.M.; Rader, A.G.; Compeer, B.; Ribeiro, C.M.S. Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses 2021, 13, 320. [Google Scholar] [CrossRef]
- Cloherty, A.P.M.; van Teijlingen, N.H.; Eisden, T.-J.T.H.D.; van Hamme, J.L.; Rader, A.G.; Geijtenbeek, T.B.H.; Schreurs, R.R.C.E.; Ribeiro, C.M.S. Autophagy-Enhancing Drugs Limit Mucosal HIV-1 Acquisition and Suppress Viral Replication Ex Vivo. Sci. Rep. 2021, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and Autophagic Cell Death: The Dark Side of Autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Luk, B.T.; Wei, X.; Campbell, G.R.; Fang, R.H.; Zhang, L.; Spector, S.A. Selective Cell Death of Latently HIV-Infected CD4+ T Cells Mediated by Autosis Inducing Nanopeptides. Cell Death Dis. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Li, Q.; Lee, J.-Y.; Lee, S.-H.; Jeong, J.H.; Lee, H.-R.; Chang, H.; Zhou, F.-C.; Gao, S.-J.; Liang, C.; et al. FLIP-Mediated Autophagy Regulation in Cell Death Control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, W.; Bauch, T.; Graviss, E.A.; Arduino, R.C.; Kimata, J.T.; Chen, M.; Wang, J. Clearance of HIV Infection by Selective Elimination of Host Cells Capable of Producing HIV. Nat. Commun. 2020, 11, 4051. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, M.; Budai, M.M.; Rice, A.P.; Kimata, J.T.; Mohan, M.; Wang, J. Clearance of HIV-1 or SIV Reservoirs by Promotion of Apoptosis and Inhibition of Autophagy: Targeting Intracellular Molecules in Cure-Directed Strategies. J. Leukoc. Biol. 2022, 112, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Metur, S.P.; Klionsky, D.J. Adaptive Immunity at the Crossroads of Autophagy and Metabolism. Cell Mol. Immunol. 2021, 18, 1096–1105. [Google Scholar] [CrossRef]
- Massanella, M.; Fromentin, R.; Chomont, N. Residual Inflammation and Viral Reservoirs: Alliance against an HIV Cure. Curr. Opin. HIV AIDS 2016, 11, 234–241. [Google Scholar] [CrossRef]
- Judith, D.; Versapuech, M.; Bejjani, F.; Palaric, M.; Verlhac, P.; Kuster, A.; Lepont, L.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. ATG5 Selectively Engages Virus-Tethered BST2/Tetherin in an LC3C-Associated Pathway. Proc. Natl. Acad. Sci. USA 2023, 120, e2217451120. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klute, S.; Sparrer, K.M.J. Friends and Foes: The Ambivalent Role of Autophagy in HIV-1 Infection. Viruses 2024, 16, 500. https://doi.org/10.3390/v16040500
Klute S, Sparrer KMJ. Friends and Foes: The Ambivalent Role of Autophagy in HIV-1 Infection. Viruses. 2024; 16(4):500. https://doi.org/10.3390/v16040500
Chicago/Turabian StyleKlute, Susanne, and Konstantin M. J. Sparrer. 2024. "Friends and Foes: The Ambivalent Role of Autophagy in HIV-1 Infection" Viruses 16, no. 4: 500. https://doi.org/10.3390/v16040500
APA StyleKlute, S., & Sparrer, K. M. J. (2024). Friends and Foes: The Ambivalent Role of Autophagy in HIV-1 Infection. Viruses, 16(4), 500. https://doi.org/10.3390/v16040500