
The Inhibition of Gag-Pol Expression by the Restriction Factor Shiftless Is Dispensable for the Restriction of HIV-1 Infection

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. Generation of HIV/MLV Single-Cycle Particles and Transduction of Target Cells
2.4. Production of HIV-1 NL4-3 and Infection of TZM-bl Reporter Cells
2.5. Analysis of HIV/MLV Gag-Pol Expression by SDS-PAGE and Immunoblotting
2.6. Analysis of SFL Interactions via Co-Immunoprecipitation
2.7. Ribosome Pelleting Assay
2.8. mRNA Binding Assay
2.9. Statistical Analysis
3. Results
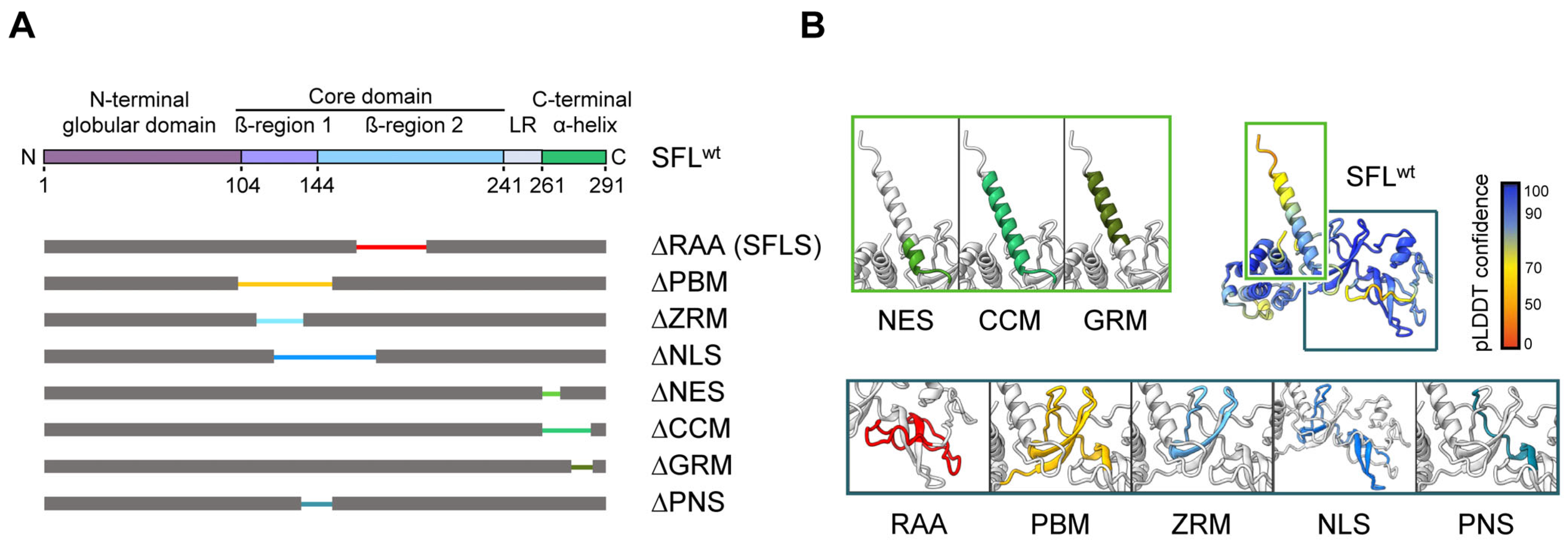
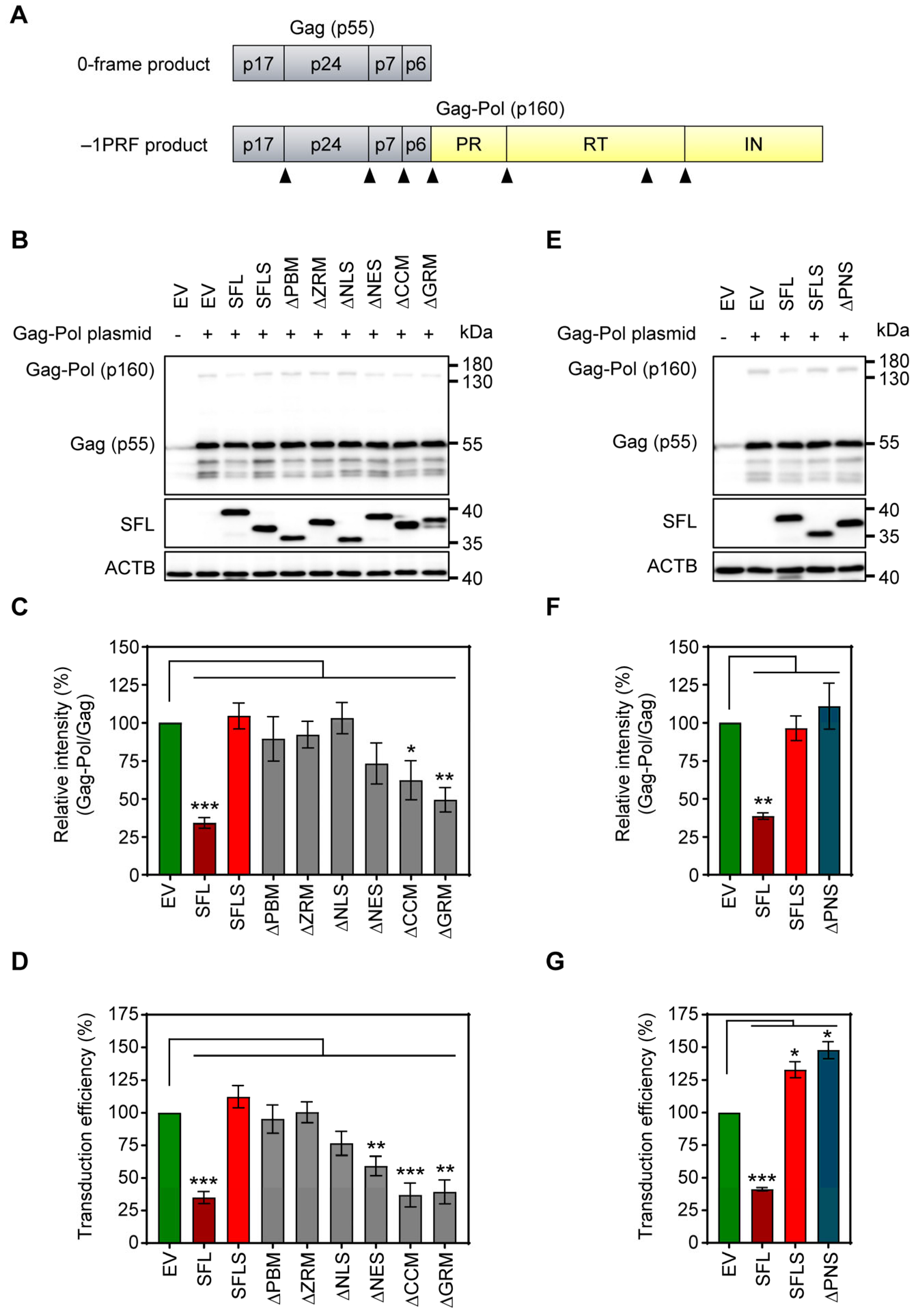
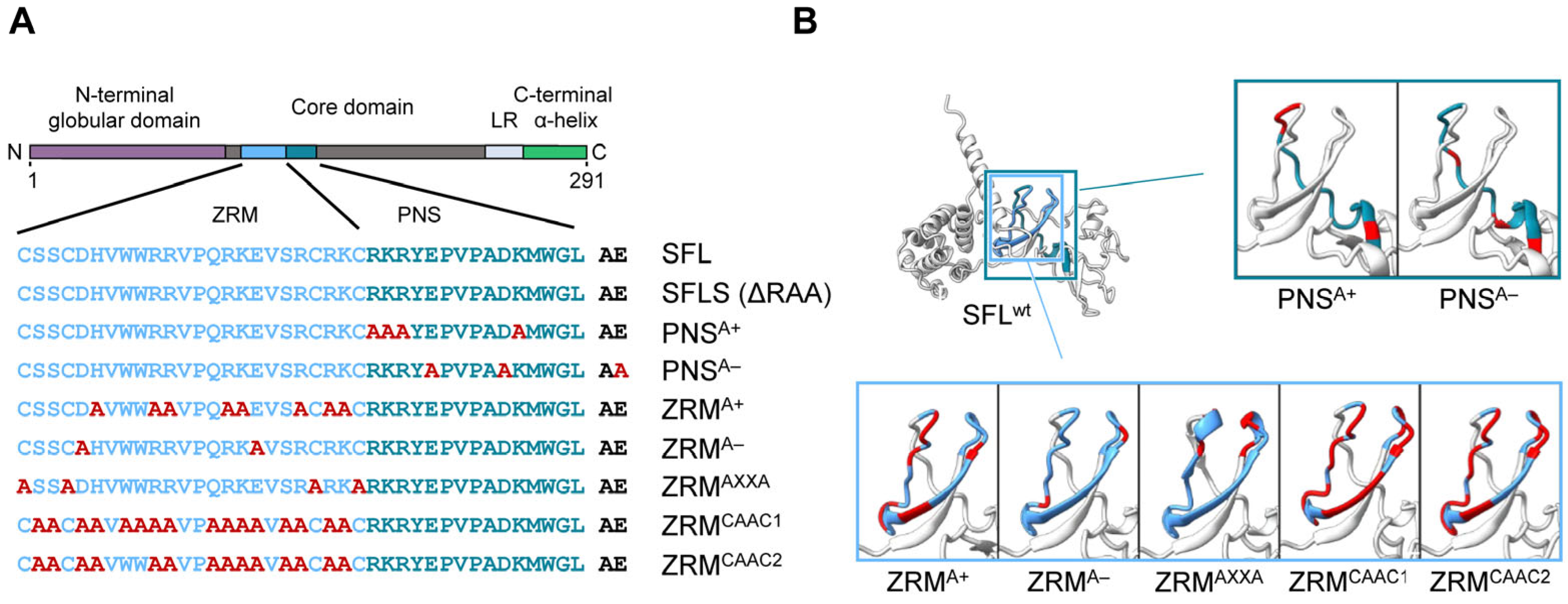
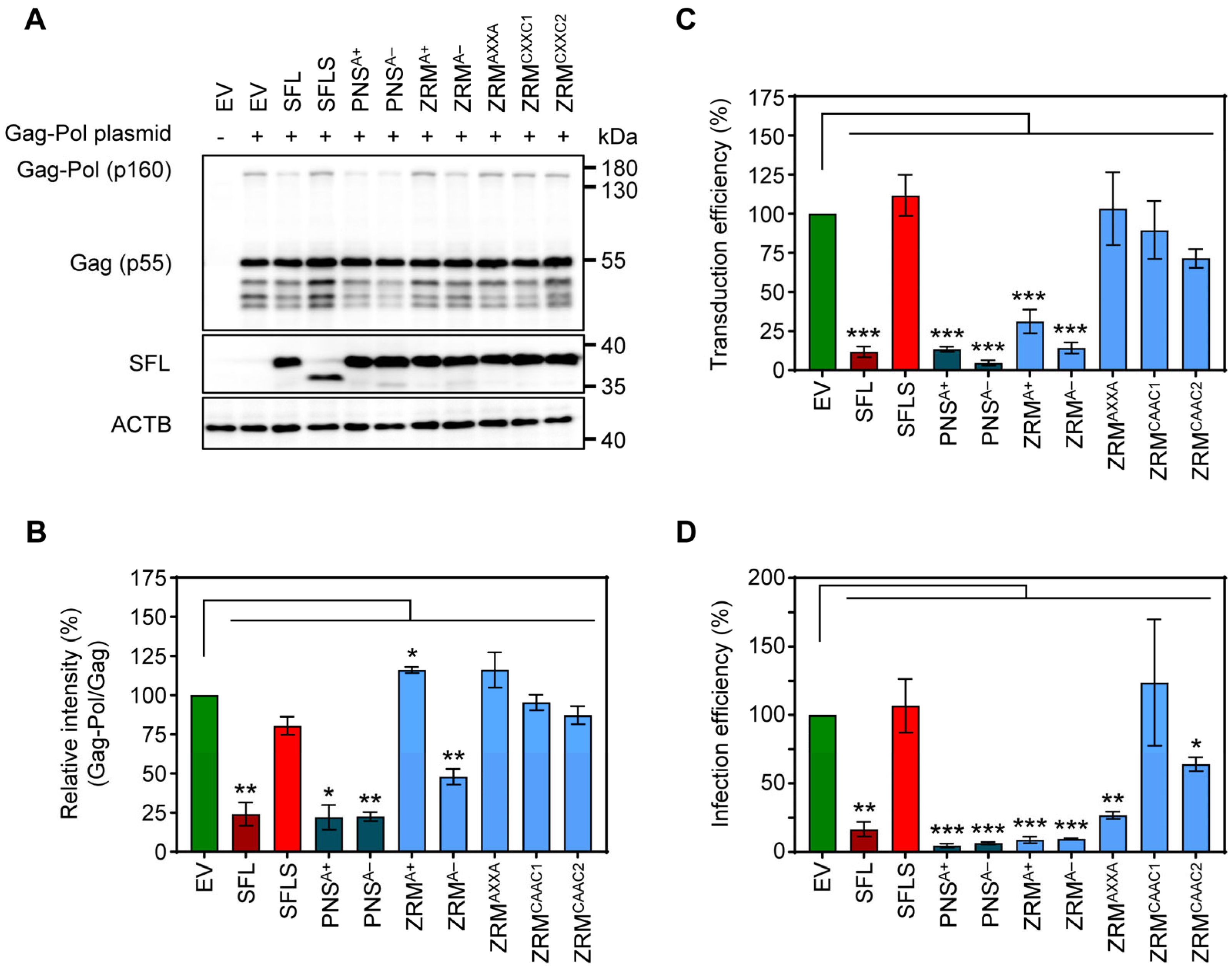
3.1. Deletion of the ZRM and PNS Regions of SFL Suppresses Gag-Pol Expression and Antiviral Activity
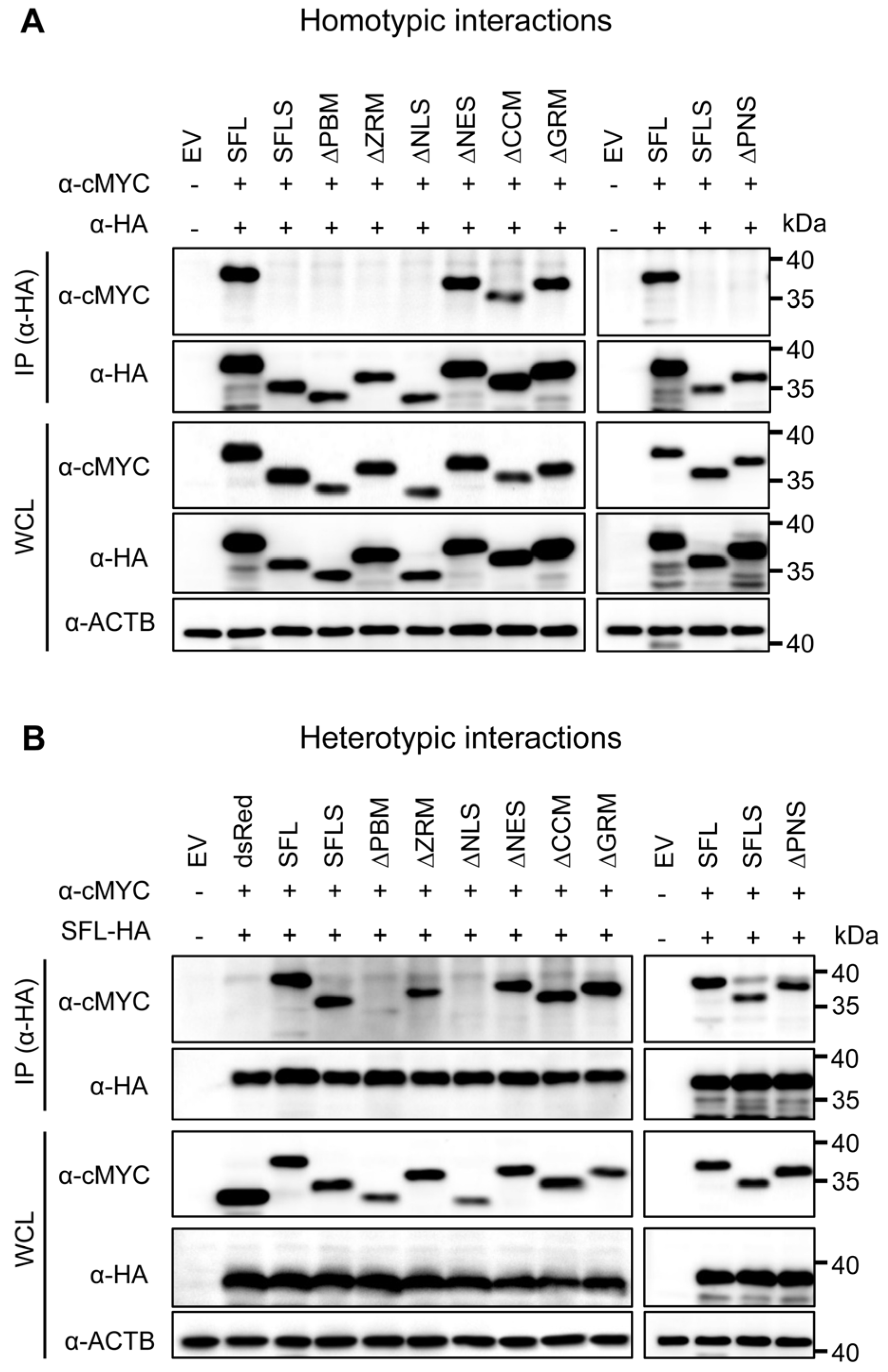
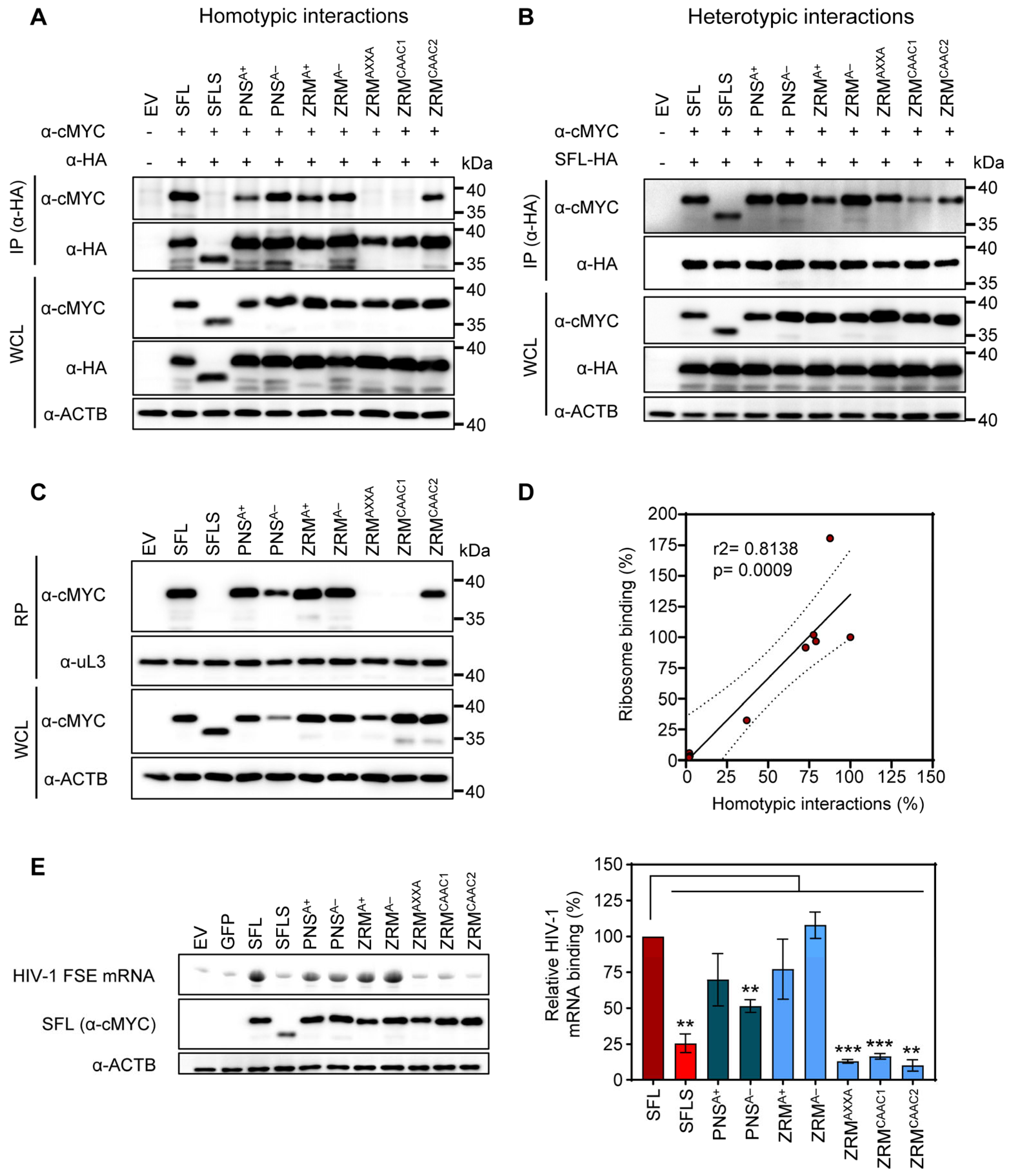
3.2. The Structural Integrity of the β-Region 1 Is Required for SFL Multimerization
3.3. Inhibition of Gag-Pol Expression and HIV-1 Infectivity Are Separable Traits of SFL
3.4. Self-Interactions, Ribosome and HIV-1 RNA Binding Are Dispensable for SFL Antiviral Activity
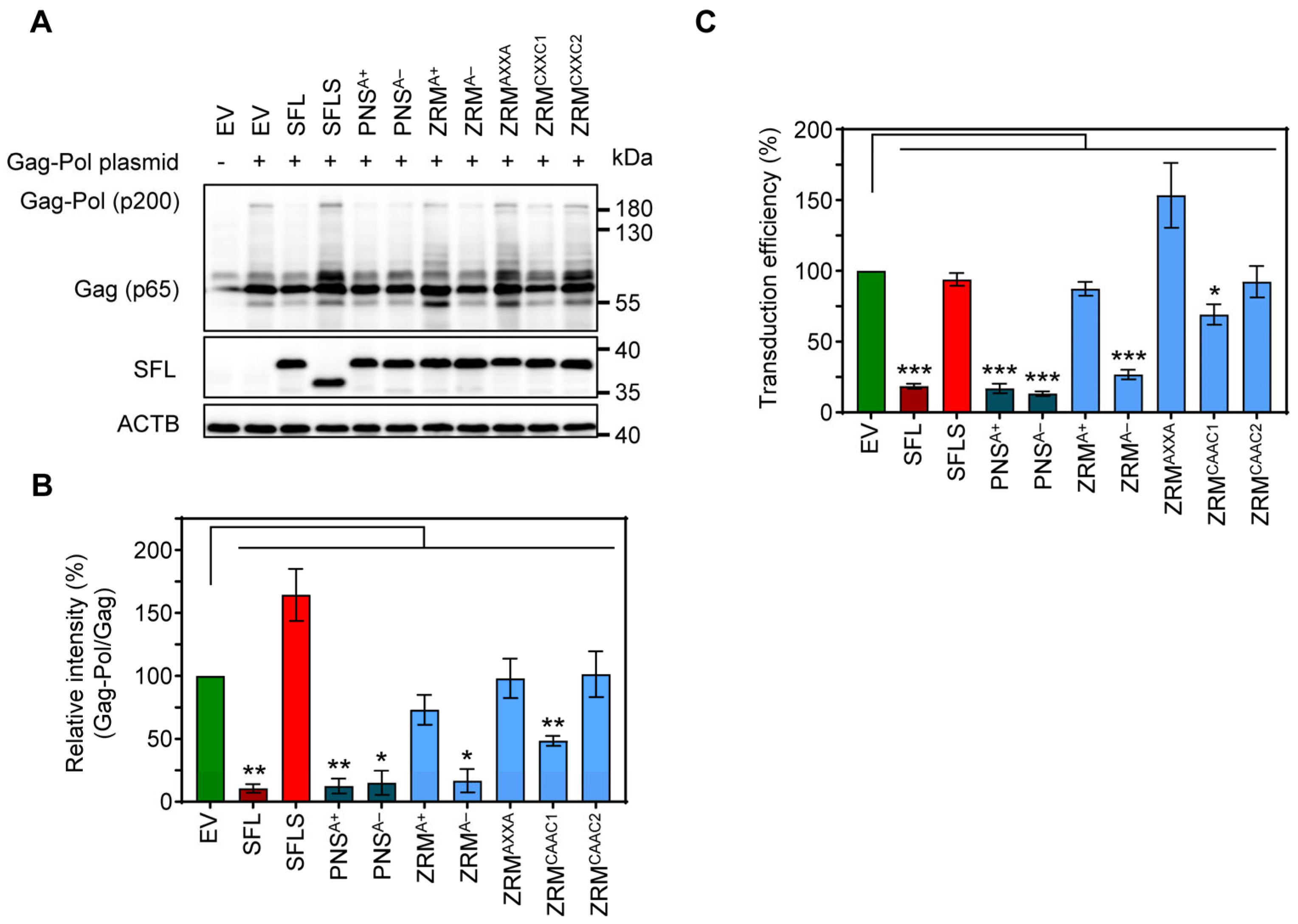
3.5. SFL Inhibits MLV Gag-Pol Expression and the Formation of Single-Cycle MLV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Noser, J.A.; Ohmine, S.; Ikeda, Y. Inhibition of HIV-1 replication by simian restriction factors, TRIM5alpha and APOBEC3G. Gene Ther. 2007, 14, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed-1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e14. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, D.; Therrien, R.; Kettaf, N.; Angermann, B.R.; Boucher, G.; Filali-Mouhim, A.; Moser, J.M.; Mehta, R.S.; Drake, D.R., 3rd; Castro, E.; et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 2008, 205, 3119–3131. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Muhlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.D.; Katze, M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: Increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nikrad, M.P.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Edeen, K.; Travanty, E.A.; Kosmider, B.; Hartshorn, K.; et al. Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Giedroc, D.P.; Cornish, P.V. Frameshifting RNA pseudoknots: Structure and mechanism. Virus Res. 2009, 139, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, N.; Peske, F.; Rodnina, M.V. Changed in translation: mRNA recoding by -1 programmed ribosomal frameshifting. Trends Biochem. Sci. 2015, 40, 265–274. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Korniy, N.; Klimova, M.; Karki, P.; Peng, B.Z.; Senyushkina, T.; Belardinelli, R.; Maracci, C.; Wohlgemuth, I.; Samatova, E.; et al. Translational recoding: Canonical translation mechanisms reinterpreted. Nucleic Acids Res. 2020, 48, 1056–1067. [Google Scholar] [CrossRef]
- Korniy, N.; Samatova, E.; Anokhina, M.M.; Peske, F.; Rodnina, M.V. Mechanisms and biomedical implications of -1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 2019, 593, 1468–1482. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed]
- Advani, V.M.; Dinman, J.D. Reprogramming the genetic code: The emerging role of ribosomal frameshifting in regulating cellular gene expression. Bioessays 2016, 38, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef] [PubMed]
- Jager, N.; Ayyub, S.A.; Korniy, N.; Peske, F.; Hoffmann, M.; Rodnina, M.V.; Pohlmann, S. Mutagenic Analysis of the HIV Restriction Factor Shiftless. Viruses 2022, 14, 1454. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.L.; Perera, L.; Blackshear, P.J. An Ancient Family of RNA-Binding Proteins: Still Important! Trends Biochem. Sci. 2017, 42, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, C.; Hoffmann, M.; Lubke, A.; Nehlmeier, I.; Kramer-Kuhl, A.; Winkler, M.; Pohlmann, S. The glycoprotein of vesicular stomatitis virus promotes release of virus-like particles from tetherin-positive cells. PLoS ONE 2017, 12, e0189073. [Google Scholar] [CrossRef]
- Ciesek, S.; Westhaus, S.; Wicht, M.; Wappler, I.; Henschen, S.; Sarrazin, C.; Hamdi, N.; Abdelaziz, A.I.; Strassburg, C.P.; Wedemeyer, H.; et al. Impact of intra- and interspecies variation of occludin on its function as coreceptor for authentic hepatitis C virus particles. J. Virol. 2011, 85, 7613–7621. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Korniy, N.; Goyal, A.; Hoffmann, M.; Samatova, E.; Peske, F.; Pohlmann, S.; Rodnina, M.V. Modulation of HIV-1 Gag/Gag-Pol frameshifting by tRNA abundance. Nucleic Acids Res. 2019, 47, 5210–5222. [Google Scholar] [CrossRef] [PubMed]
- Balke, D.; Frommer, J.; Rublack, N.; Springstubbe, D.; Appel, B.; Müller, S. Synthesis of Site-Specifically Modified Long-mer RNAs. In Chemical Biology of Nucleic Acids, RNA Technologies; Erdmann, V., Markiewicz, W., Barciszewski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Milon, P.; Konevega, A.L.; Peske, F.; Fabbretti, A.; Gualerzi, C.O.; Rodnina, M.V. Transient kinetics, fluorescence, and FRET in studies of initiation of translation in bacteria. Methods Enzymol. 2007, 430, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Proudnikov, D.; Mirzabekov, A. Chemical methods of DNA and RNA fluorescent labeling. Nucleic Acids Res. 1996, 24, 4535–4542. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.R.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Gupta, A.; Gribskov, M. The role of RNA sequence and structure in RNA—Protein interactions. J. Mol. Biol. 2011, 409, 574–587. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Khrapov, M.A.; Cox, J.C.; Yao, J.; Tong, L.; Ellington, A.D. AANT: The Amino Acid-Nucleotide Interaction Database. Nucleic Acids Res. 2004, 32, D174–D181. [Google Scholar] [CrossRef] [PubMed]
- Treger, M.; Westhof, E. Statistical analysis of atomic contacts at RNA-protein interfaces. J. Mol. Recognit. 2001, 14, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Gendron, K.; Dulude, D.; Lemay, G.; Ferbeyre, G.; Brakier-Gingras, L. The virion-associated Gag-Pol is decreased in chimeric Moloney murine leukemia viruses in which the readthrough region is replaced by the frameshift region of the human immunodeficiency virus type 1. Virology 2005, 334, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Collins, J.T.; D’Souza, V.M.; Telesnitsky, A. Determinants of Moloney murine leukemia virus Gag-Pol and genomic RNA proportions. J. Virol. 2014, 88, 7267–7275. [Google Scholar] [CrossRef] [PubMed]
- Houck-Loomis, B.; Durney, M.A.; Salguero, C.; Shankar, N.; Nagle, J.M.; Goff, S.P.; D’Souza, V.M. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 2011, 480, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, M.N.; Brakier-Gingras, L.; Lemay, G. Replacement of murine leukemia virus readthrough mechanism by human immunodeficiency virus frameshift allows synthesis of viral proteins and virus replication. J. Virol. 2003, 77, 3345–3350. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.; Muller, M. Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses 2022, 14, 1338. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Chin, W.X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Biancalana, M.; Grant, J.C.; Chiu, H.J.; Jaroszewski, L.; Knuth, M.W.; Lesley, S.A.; Godzik, A.; Elsliger, M.A.; Deacon, A.M.; et al. Structures of single-layer beta-sheet proteins evolved from beta-hairpin repeats. Protein Sci. 2019, 28, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Balinsky, C.A.; Schmeisser, H.; Wells, A.I.; Ganesan, S.; Jin, T.; Singh, K.; Zoon, K.C. IRAV (FLJ11286), an Interferon-Stimulated Gene with Antiviral Activity against Dengue Virus, Interacts with MOV10. J. Virol. 2017, 91, e01606-16. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM proteins inhibit HIV-1 protein synthesis. Sci. Rep. 2018, 8, 14551. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Polonis, V.R.; Brown, B.K.; Rosa Borges, A.; Zolla-Pazner, S.; Dimitrov, D.S.; Zhang, M.Y.; Barnett, S.W.; Ruprecht, R.M.; Scarlatti, G.; Fenyo, E.M.; et al. Recent advances in the characterization of HIV-1 neutralization assays for standardized evaluation of the antibody response to infection and vaccination. Virology 2008, 375, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, M.; Parkin, N.; Varmus, H.E. An RNA pseudoknot and an optimal heptameric shift site are required for highly efficient ribosomal frameshifting on a retroviral messenger RNA. Proc. Natl. Acad. Sci. USA 1992, 89, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.F.; Crowe-McAuliffe, C.; Graves, R.; Cardno, T.S.; McKinney, C.; Poole, E.S.; Tate, W.P. The highly conserved codon following the slippery sequence supports -1 frameshift efficiency at the HIV-1 frameshift site. PLoS ONE 2015, 10, e0122176. [Google Scholar] [CrossRef]
- Csibra, E.; Brierley, I.; Irigoyen, N. Modulation of stop codon read-through efficiency and its effect on the replication of murine leukemia virus. J. Virol. 2014, 88, 10364–10376. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, N.; Dinan, A.M.; Brierley, I.; Firth, A.E. Ribosome profiling of the retrovirus murine leukemia virus. Retrovirology 2018, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaka, Y.; Katoh, I.; Copeland, T.D.; Oroszlan, S. Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc. Natl. Acad. Sci. USA 1985, 82, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Napthine, S.; Hill, C.H.; Nugent, H.C.M.; Brierley, I. Modulation of Viral Programmed Ribosomal Frameshifting and Stop Codon Readthrough by the Host Restriction Factor Shiftless. Viruses 2021, 13, 1230. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.A.; Dinman, J.D. Shiftless Is a Novel Member of the Ribosome Stress Surveillance Machinery That Has Evolved to Play a Role in Innate Immunity and Cancer Surveillance. Viruses 2023, 15, 2296. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jäger, N.; Ayyub, S.A.; Peske, F.; Liedtke, D.; Bohne, J.; Hoffmann, M.; Rodnina, M.V.; Pöhlmann, S. The Inhibition of Gag-Pol Expression by the Restriction Factor Shiftless Is Dispensable for the Restriction of HIV-1 Infection. Viruses 2024, 16, 583. https://doi.org/10.3390/v16040583
Jäger N, Ayyub SA, Peske F, Liedtke D, Bohne J, Hoffmann M, Rodnina MV, Pöhlmann S. The Inhibition of Gag-Pol Expression by the Restriction Factor Shiftless Is Dispensable for the Restriction of HIV-1 Infection. Viruses. 2024; 16(4):583. https://doi.org/10.3390/v16040583
Chicago/Turabian StyleJäger, Niklas, Shreya Ahana Ayyub, Frank Peske, David Liedtke, Jens Bohne, Markus Hoffmann, Marina V. Rodnina, and Stefan Pöhlmann. 2024. "The Inhibition of Gag-Pol Expression by the Restriction Factor Shiftless Is Dispensable for the Restriction of HIV-1 Infection" Viruses 16, no. 4: 583. https://doi.org/10.3390/v16040583
APA StyleJäger, N., Ayyub, S. A., Peske, F., Liedtke, D., Bohne, J., Hoffmann, M., Rodnina, M. V., & Pöhlmann, S. (2024). The Inhibition of Gag-Pol Expression by the Restriction Factor Shiftless Is Dispensable for the Restriction of HIV-1 Infection. Viruses, 16(4), 583. https://doi.org/10.3390/v16040583