Molecular Basis for Drug Resistance in HIV-1 Protease

Abstract
:1. Introduction
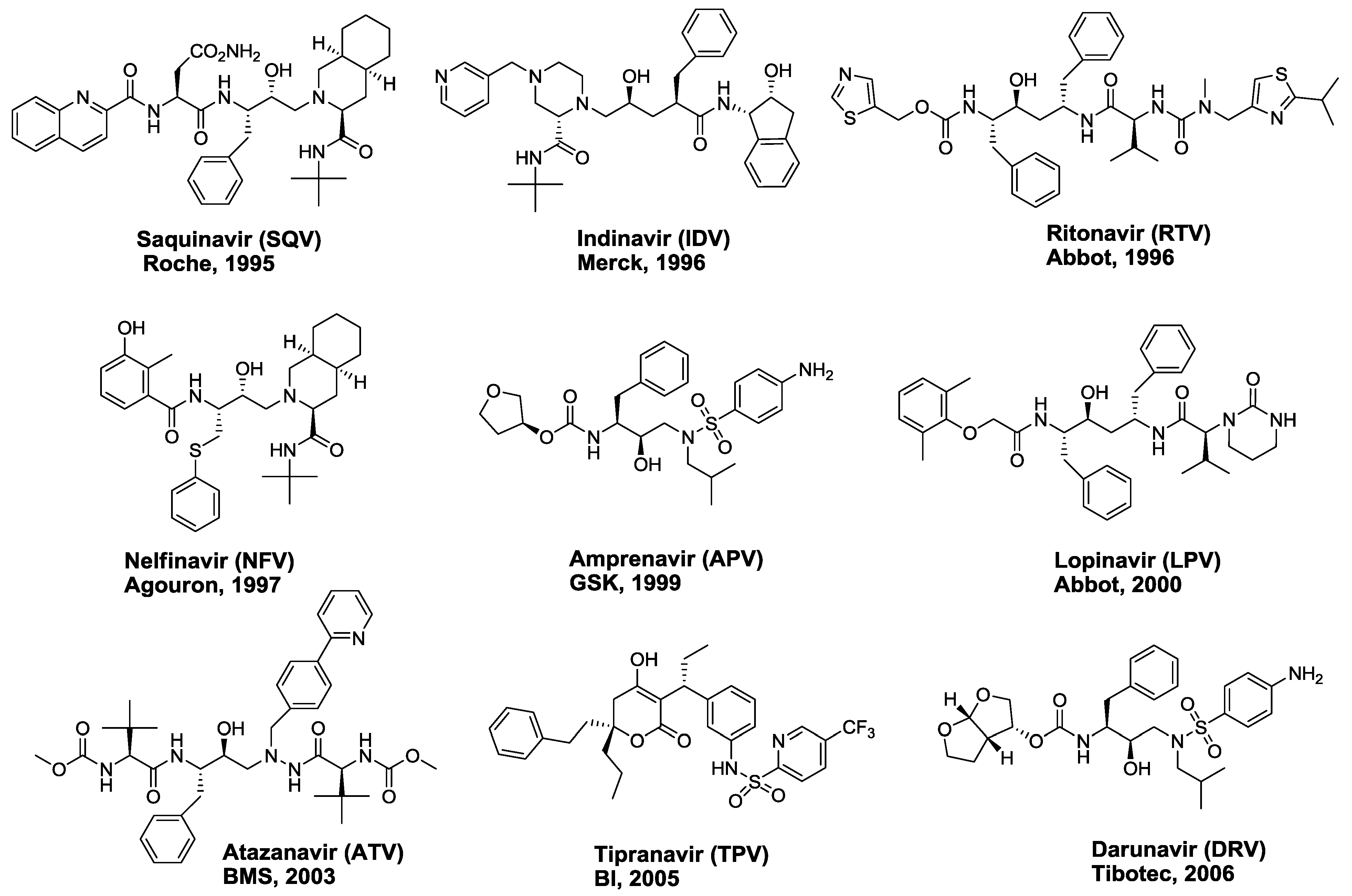
2. FDA-approved HIV-1 Protease Inhibitors
3. Interdependency of Drug Resistance
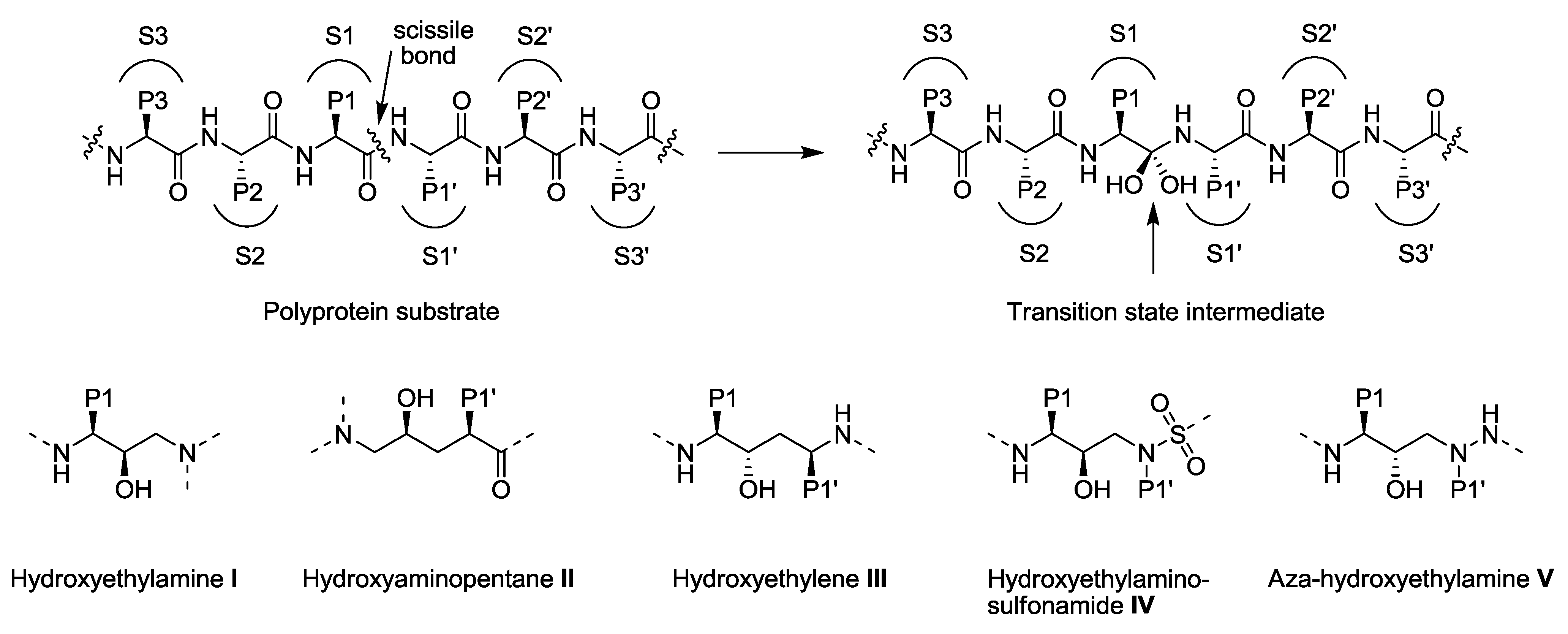
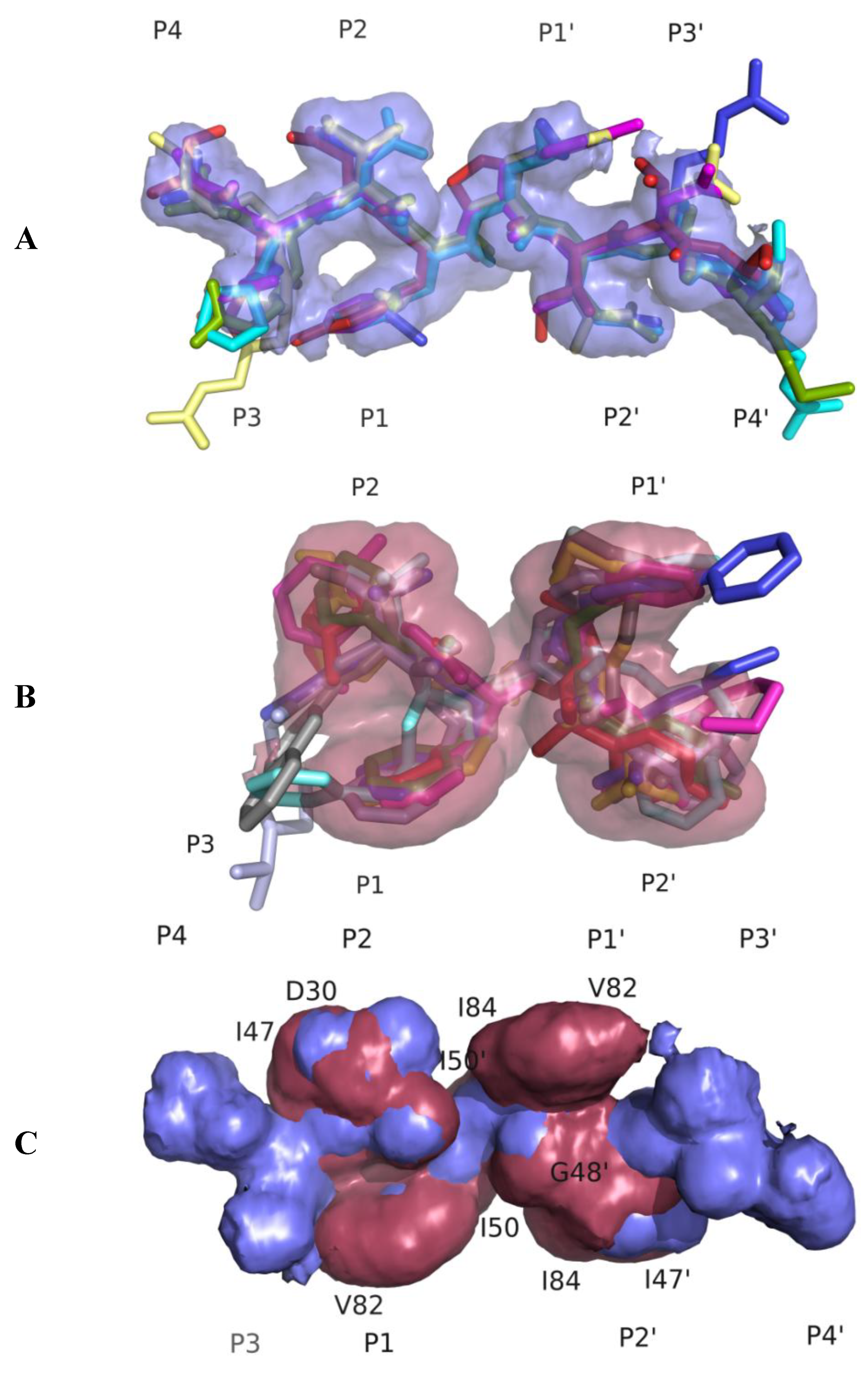
3.1. Substrate Envelope Hypotheses
3.2. Drug Resistance—A Change in Molecular Recognition at the Active Site
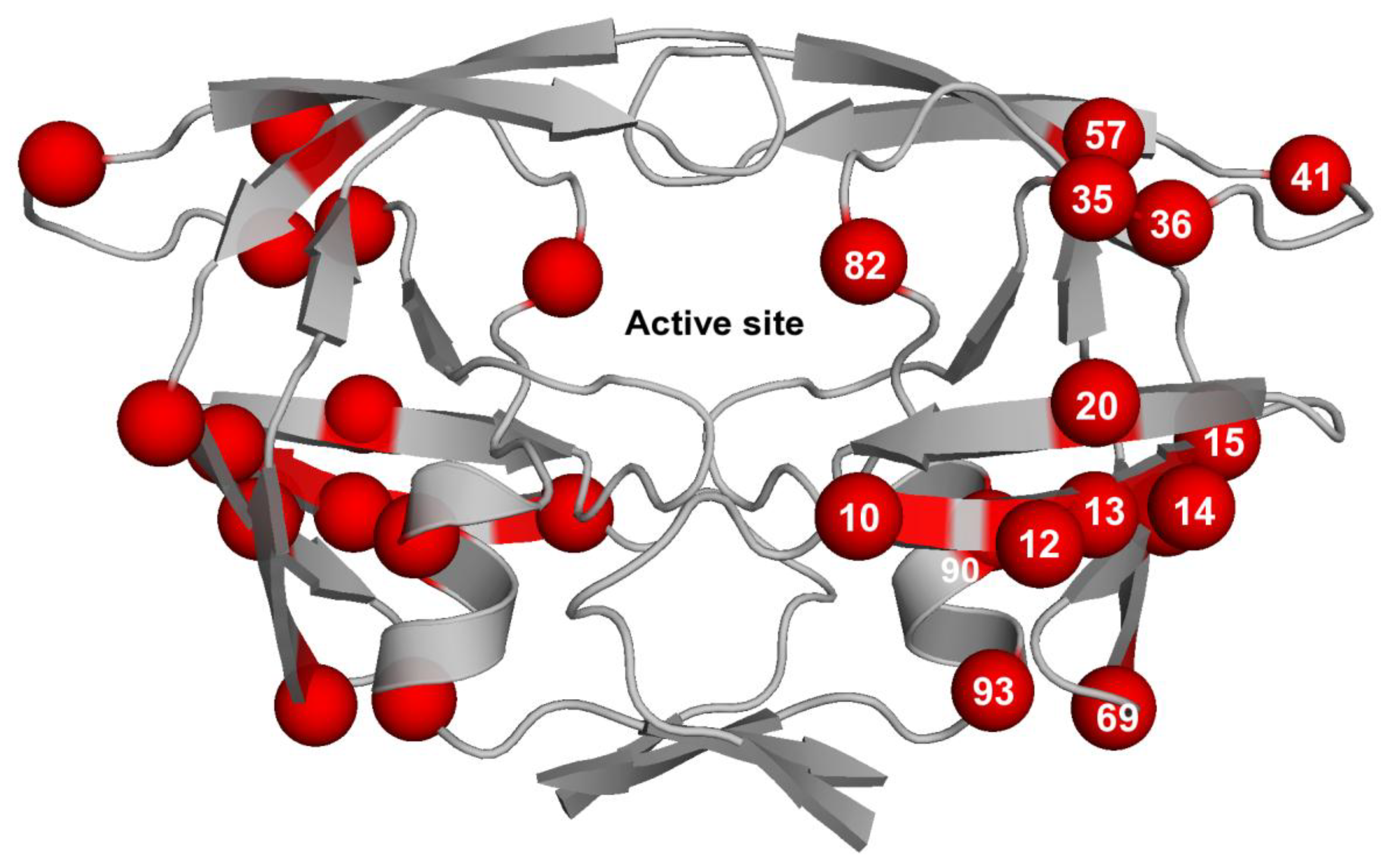
3.3. Contribution of Protease Mutations outside the Active Site
3.4. Impact of the Co-evolution of Protease Cleavage Sites on Resistance
4. Altered Pathways to Drug Resistance between the HIV-1 Clades
5. The Atomic Energetics of Drug Resistance
6. Incorporating the Substrate Envelope Constraint in Structure Based Drug Design
7. Conclusions
Acknowledgments
References and Notes
- The Joint United Nations Program on HIV/AIDS (UNAIDS). 2008 Report on the Global AIDS Epidemic; UNAIDS/08.25E/JC1510E; UNAIDS: Geneva, Switzerland, 2008.
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus drug resistance: An update. Antivir. Res. 2010, 85, 210–231. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cajas, J.L.; Wainberg, M.A. Protease inhibitor resistance in HIV-infected patients: Molecular and clinical perspectives. Antivir. Res. 2007, 76, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
- Mehellou, Y.; De Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Krohn, A.; Lambert, R.W.; Merrett, J.H.; Mills, J.S.; Parkes, K.E.B.; Redshaw, S.; Ritchie, A.J.; Taylor, D.L.; Thomas, G.J.; Machin, P.J. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, B.D.; Levin, R.B.; McDaniel, S.L.; Vacca, J.P.; Guare, J.P.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; Schleif, W.A.; Quintero, J.C.; Lin, J.H.; Chen, I.-W.; Holloway, M.K.; Fitzgerald, P.M.D.; Axel, M.G.; Ostovic, D.; Anderson, P.S.; Huff, J.R. L-735,524: The design of a potent and orally bioavailable HIV protease inhibitor. J. Med. Chem. 1994, 37, 3443–3451. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.P.; Wideburg, N.E.; Saldivar, A.; Ruiz, L.; Kati, W.M.; Sham, H.L.; Robins, T.; Stewart, K.D.; Hsu, A.; Plattner, J.J.; Leonard, J.M.; Norbeck, D.W. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2484–2488. [Google Scholar] [CrossRef] [PubMed]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F., 2nd; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; Dressman, B.A.; Hatch, S.D.; Khalil, D.A.; Kosa, M.B.; Lubbehusen, P.P.; Muesing, M.A.; Patick, A.K.; Reich, S.H.; Su, K.S.; Tatlock, J.H. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Kim, E.E.; Baker, C.T.; Dwyer, M.D.; Murcko, M.A.; Rao, B.G.; Tung, R.D.; Navia, M.A. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 1995, 117, 1181–1182. [Google Scholar] [CrossRef]
- Sham, H.L.; Kempf, D.J.; Molla, A.; Marsh, K.C.; Kumar, G.N.; Chen, C.M.; Kati, W.; Stewart, K.; Lal, R.; Hsu, A.; Betebenner, D.; Korneyeva, M.; Vasavanonda, S.; McDonald, E.; Saldivar, A.; Wideburg, N.; Chen, X.; Niu, P.; Park, C.; Jayanti, V.; Grabowski, B.; Granneman, G.R.; Sun, E.; Japour, A.J.; Leonard, J.M.; Plattner, J.J.; Norbeck, D.W. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.S.; Riccardi, K.A.; Gong, Y.F.; Guo, Q.; Stock, D.A.; Blair, W.S.; Terry, B.J.; Deminie, C.A.; Djang, F.; Colonno, R.J.; Lin, P.F. BMS-232632, a highly potent human immunodeficiency virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob. Agents Chemother. 2000, 44, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.R.; Strohbach, J.W.; Tommasi, R.A.; Aristoff, P.A.; Johnson, P.D.; Skulnick, H.I.; Dolak, L.A.; Seest, E.P.; Tomich, P.K.; Bohanon, M.J.; Horng, M.M.; Lynn, J.C.; Chong, K.T.; Hinshaw, R.R.; Watenpaugh, K.D.; Janakiraman, M.N.; Thaisrivongs, S. Tipranavir (PNU-140690): A potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J. Med. Chem. 1998, 41, 3467–3476. [Google Scholar] [CrossRef]
- De Meyer, S.; Azijn, H.; Surleraux, D.; Jochmans, D.; Tahri, A.; Pauwels, R.; Wigerinck, P.; de Bethune, M.P. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 2005, 49, 2314–2321. [Google Scholar] [CrossRef]
- Koh, Y.; Nakata, H.; Maeda, K.; Ogata, H.; Bilcer, G.; Devasamudram, T.; Kincaid, J.F.; Boross, P.; Wang, Y.F.; Tie, Y.; Volarath, P.; Gaddis, L.; Harrison, R.W.; Weber, I.T.; Ghosh, A.K.; Mitsuya, H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2003, 47, 3123–3129. [Google Scholar] [CrossRef]
- Surleraux, D.L.; Tahri, A.; Verschueren, W.G.; Pille, G.M.; de Kock, H.A.; Jonckers, T.H.; Peeters, A.; De Meyer, S.; Azijn, H.; Pauwels, R.; de Bethune, M.P.; King, N.M.; Prabu-Jeyabalan, M.; Schiffer, C.A.; Wigerinck, P.B. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem. 2005, 48, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Meibohm, A.; Condra, J.H.; Valentine, F.T.; McMahon, D.; Gonzalez, C.; Jonas, L.; Emini, E.A.; Chodakewitz, J.A.; Isaacs, R.; Richman, D.D. 3-Year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann. Intern. Med. 2000, 133, 35–39. [Google Scholar] [CrossRef]
- Bartlett, J.A.; DeMasi, R.; Quinn, J.; Moxham, C.; Rousseau, F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS 2001, 15, 1369–1377. [Google Scholar] [CrossRef]
- Palella, F.J.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D.; The, H.I.V.O.S.I. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef]
- Hogg, R.S.; Heath, K.V.; Yip, B.; Craib, K.J.P.; O'Shaughnessy, M.V.; Schechter, M.T.; Montaner, J.S.G. Improved survival among HIV-infected individuals following initiation of antiretroviral therapy. JAMA 1998, 279, 450–454. [Google Scholar] [CrossRef]
- Zeldin, R.K.; Petruschke, R.A. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J. Antimicrob. Chemother. 2004, 53, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Gulnik, S.V.; Eissenstat, M. Approaches to the design of HIV protease inhibitors with improved resistance profiles. Curr. Opin. HIV AIDS 2008, 3, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Nalam, M.N.L.; Schiffer, C.A. New approaches to HIV protease inhibitor drug design II: Testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr. Opin. HIV AIDS 2008, 3, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2008, 41, 78–86. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Leshchenko-Yashchuk, S.; Anderson, D.D.; Baldridge, A.; Noetzel, M.; Miller, H.B.; Tie, Y.; Wang, Y.F.; Koh, Y.; Weber, I.T.; Mitsuya, H. Design of HIV-1 protease inhibitors with pyrrolidinones and oxazolidinones as novel P1'-ligands to enhance backbone-binding interactions with protease: Synthesis, biological evaluation, and protein-ligand X-ray studies. J. Med. Chem. 2009, 52, 3902–3914. [Google Scholar] [CrossRef]
- Cihlar, T.; He, G.X.; Liu, X.; Chen, J.M.; Hatada, M.; Swaminathan, S.; McDermott, M.J.; Yang, Z.Y.; Mulato, A.S.; Chen, X.; Leavitt, S.A.; Stray, K.M.; Lee, W.A. Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol. 2006, 363, 635–647. [Google Scholar] [CrossRef]
- Stranix, B.R.; Sauve, G.; Bouzide, A.; Cote, A.; Sevigny, G.; Yelle, J. Lysine sulfonamides as novel HIV-protease inhibitors: Optimization of the Nepsilon-acyl-phenyl spacer. Bioorg. Med. Chem. Lett. 2003, 13, 4289–4292. [Google Scholar] [CrossRef]
- Altman, M.D.; Ali, A.; Reddy, G.S.K.K.; Nalam, M.N.L.; Anjum, S.G.; Cao, H.; Chellappan, S.; Kairys, V.; Fernandes, M.X.; Gilson, M.K.; Schiffer, C.A.; Rana, T.M.; Tidor, B. HIV-1 Protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J. Am. Chem. Soc. 2008, 130, 6099–6113. [Google Scholar] [CrossRef]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef]
- Navia, M.A.; Fitzgerald, P.M.D.; McKeever, B.M.; Leu, C.-T.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Miller, M.; Schneider, J.; Sathyanarayana, B.K.; Toth, M.V.; Marshall, G.R.; Clawson, L.; Selk, L.; Kent, S.B.; Wlodawer, A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2. 3 A resolution. Science 1989, 246, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; Sun, E.; Pizzuti, D.; Plattner, J.J.; Norbeck, D.W.; Leonard, J.M. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [CrossRef]
- Nalam, M.N.L.; Ali, A.; Altman, M.D.; Reddy, G.S.K.K.; Chellappan, S.; Kairys, V.; Ozen, A.; Cao, H.; Gilson, M.K.; Tidor, B.; Rana, T.M.; Schiffer, C.A. Evaluating the substrate-envelope hypothesis: Structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J. Virol. 2010, 84, 5368–5378. [Google Scholar] [CrossRef] [PubMed]
- Surleraux, D.L.; de Kock, H.A.; Verschueren, W.G.; Pille, G.M.; Maes, L.J.; Peeters, A.; Vendeville, S.; De Meyer, S.; Azijn, H.; Pauwels, R.; de Bethune, M.P.; King, N.M.; Prabu-Jeyabalan, M.; Schiffer, C.A.; Wigerinck, P.B. Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J. Med. Chem. 2005, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- King, N.M.; Prabu-Jeyabalan, M.; Nalivaika, E.A.; Schiffer, C.A. Combating susceptibility to drug resistance: Lessons from HIV-1 protease. Chem. Biol. 2004, 11, 1333–1338. [Google Scholar]
- King, N. M.; Prabu-Jeyabalan, M.; Nalivaika, E. A.; Wigerinck, P.; de Bethune, M. P.; Schiffer, C. A. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J. Virol. 2004, 78, 12012–12021. [Google Scholar] [CrossRef]
- Lefebvre, E.; Schiffer, C.A. Resilience to resistance of HIV-1 protease inhibitors: Profile of darunavir. AIDS Rev. 2008, 10, 131–142. [Google Scholar]
- Stanford HIV Drug Resistance Database. Available online: http://hivdb.Stanford.edu (accessed on 20 October 2010).
- Wu, T.D.; Schiffer, C.A.; Gonzales, M.J.; Taylor, J.; Kantor, R.; Chou, S.; Israelski, D.; Zolopa, A.R.; Fessel, W.J.; Shafer, R.W. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J. Virol. 2003, 77, 4836–4847. [Google Scholar] [CrossRef]
- Gulnik, S.V.; Suvorov, L.I.; Liu, B.; Yu, B.; Anderson, B.; Mitsuya, H.; Erickson, J.W. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 1995, 34, 9282–9287. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.; Duran, M.; Cao, Y.; Shugarts, D.; Keller, M.; Mazabel, E.; Knowles, M.; Chapman, S.; Kuritzkes, D.; Markowitz, M. Genotypic and phenotypic characterization of human immunodeficiency virus type 1 variants isolated from patients treated with the protease inhibitor nelfinavir. Antimicrob. Agents Chemother. 1998, 42, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, B.; Louis, J.; Reed, C.; Adomat, J.; Krouse, J.; Wang, Y.; Harrison, R.; Weber, I. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur. J. Biochem. 1999, 263, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Colonno, R.; Rose, R.; McLaren, C.; Thiry, A.; Parkin, N.; Friborg, J. Identification of I50L as the signature atazanavir (ATV)-resistance mutation in treatment-naive HIV-1-infected patients receiving ATV-containing regimens. J. Infect. Dis. 2004, 189, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Grant, R.M.; Beatty, G.W.; Horton, C.; Detmer, J.; Eastman, S. Activity of a ritonavir plus saquinavir-containing regimen in patients with virologic evidence of indinavir or ritonavir failure. AIDS Res. Hum. Retroviruses 1998, 12, F97–F102. [Google Scholar] [CrossRef]
- Molla, A.; Korneyeva, M.; Gao, Q.; Vasavanonda, S.; Schipper, P.J.; Mo, H.M.; Markowitz, M.; Chernyavskiy, T.; Niu, P.; Lyons, N.; Hsu, A.; Granneman, G.R.; Ho, D.D.; Boucher, C.A.; Leonard, J.M.; Norbeck, D.W.; Kempf, D.J. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 1996, 2, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Zolopa, A.R.; Shafer, R.W.; Warford, A.; Montoya, J.G.; Hsu, P.; Katzenstein, D.; Merigan, T.C.; Efron, B. HIV-1 genotypic resistance patterns predict response to saquinavir-ritonavir therapy in patients in whom previous protease inhibitor therapy had failed. Ann. Intern. Med. 1999, 131, 813–821. [Google Scholar] [CrossRef]
- Munshi, S.; Chen, Z.; Yan, Y.; Li, Y.; Olsen, D.B.; Schock, H.B.; Galvin, B.B.; Dorsey, B.; Kuo, L.C. An alternate binding site for the P1-P3 group of a class of potent HIV-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: Structural insights for better antiviral therapy. J. Virol. 2003, 77, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Wang, Y.F.; Kovalevsky, A.Y.; Harrison, R.W.; Weber, I.T. Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters. FEBS J. 2010, 277, 3699–3714. [Google Scholar] [CrossRef]
- Saskova, K.G.; Kozisek, M.; Lepsik, M.; Brynda, J.; Rezacova, P.; Vaclavikova, J.; Kagan, R.M.; Machala, L.; Konvalinka, J. Enzymatic and structural analysis of the I47A mutation contributing to the reduced susceptibility to HIV protease inhibitor lopinavir. Protein Sci. 2008, 17, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, C.A. University of Massachusetts Medical School, Worcester, MA, USA. Unpublished work. 2010. [Google Scholar]
- Saskova, K.G.; Kozisek, M.; Rezacova, P.; Brynda, J.; Yashina, T.; Kagan, R.M.; Konvalinka, J. Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol. 2009, 83, 8810–8818. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Boross, P.I.; Wang, Y.F.; Gaddis, L.; Liu, F.; Chen, X.; Tozser, J.; Harrison, R.W.; Weber, I.T. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005, 272, 5265–5277. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia, P.; Ruane, P.; Hellinger, J.; Shirvani, V.; Zolopa, A.; Shafer, R.W. HIV-1 Protease Mutations and Protease Inhibitor Cross Resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Vega, S.; Freire, E. Amplification of the effects of drug resistance mutations by background polymorphisms in HIV-1 protease from African subtypes. Biochemistry 2002, 41, 8613–8619. [Google Scholar] [CrossRef]
- Liu, F.; Boross, P.I.; Wang, Y.F.; Tozser, J.; Louis, J.M.; Harrison, R.W.; Weber, I.T. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J. Mol. Biol. 2005, 354, 789–800. [Google Scholar] [CrossRef]
- Clemente, J.C.; Moose, R.E.; Hemrajani, R.; Whitford, L.R.; Govindasamy, L.; Reutzel, R.; McKenna, R.; Agbandje-McKenna, M.; Goodenow, M.M.; Dunn, B.M. Comparing the accumulation of active- and nonactive-site mutations in the HIV-1 protease. Biochemistry 2004, 43, 12141–12151. [Google Scholar] [CrossRef]
- Svicher, V.; Ceccherini-Silberstein, F.; Erba, F.; Santoro, M.; Gori, C.; Bellocchi, M.C.; Giannella, S.; Trotta, M.P.; Monforte, A.; Antinori, A.; Perno, C.F. Novel human immunodeficiency virus type 1 protease mutations potentially involved in resistance to protease inhibitors. Antimicrob. Agents Chemother. 2005, 49, 2015–2025. [Google Scholar] [CrossRef]
- Luque, I.; Todd, M.J.; Gomez, J.; Semo, N.; Freire, E. Molecular basis of resistance to HIV-1 protease inhibition: A plausible hypothesis. Biochemistry 1998, 37, 5791–5797. [Google Scholar] [CrossRef]
- Mahalingam, B.; Louis, J.M.; Hung, J.; Harrision, R.W.; Weber, I.T. Structural implications of drug-resistant mutants of HIV-1 protease: High-resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins 2001, 43, 455–464. [Google Scholar] [CrossRef]
- Mahalingam, B.; Boross, P.; Wang, Y.F.; Louis, J.M.; Fischer, C.C.; Tozser, J.; Harrison, R.W.; Weber, I.T. Combining mutations in HIV-1 protease to understand mechanisms of resistance. Proteins 2002, 48, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.; Winters, M.A.; Rhee, S.Y.; Merigan, T.C.; Schiffer, C.A.; Shafer, R.W. Association of a novel human immunodeficiency virus type 1 protease substrate cleft mutation, L23I, with protease inhibitor therapy and in vitro drug resistance. Antimicrob. Agents Chemother. 2004, 48, 4864–4868. [Google Scholar] [CrossRef] [PubMed]
- Skalova, T.; Dohnalek, J.; Duskova, J.; Petrokova, H.; Hradilek, M.; Soucek, M.; Konvalinka, J.; Hasek, J. HIV-1 protease mutations and inhibitor modifications monitored on a series of complexes. Structural basis for the effect of the A71V mutation on the active site. J. Med. Chem. 2006, 49, 5777–5784. [Google Scholar] [PubMed]
- Piana, S.; Carloni, P.; Rothlisberger, U. Drug resistance in HIV-1 protease: Flexibility-assisted mechanism of compensatory mutations. Protein Sci. 2002, 11, 2393–2402. [Google Scholar] [CrossRef]
- Foulkes-Murzycki, J.E.; Scout, W.R.P.; Schiffer, C.A. Hydrophobic sliding: A possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 2007, 15, 225–233. [Google Scholar] [CrossRef]
- Shuman, C.F.; Markgren, P.O.; Hamalainen, M.; Danielson, U.H. Elucidation of HIV-1 protease resistance by characterization of interaction kinetics between inhibitors and enzyme variants. Antivir. Res. 2003, 58, 235–242. [Google Scholar] [CrossRef]
- Coman, R.M.; Robbins, A.H.; Fernandez, M.A.; Gilliland, C.T.; Sochet, A.A.; Goodenow, M.M.; McKenna, R.; Dunn, B.M. The contribution of naturally occurring polymorphisms in altering the biochemical and structural characteristics of HIV-1 subtype C protease. Biochemistry 2008, 47, 731–743. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Imamichi, H.; Imamichi, T.; Lane, H.C.; Falloon, J.; Vasudevachari, M.B.; Salzman, N.P. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J. Virol. 1997, 71, 6662–6670. [Google Scholar] [CrossRef]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [CrossRef]
- Bally, F.; Martinez, R.; Peters, S.; Sudre, P.; Telenti, A. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: Clinical significance and implications for resistance to protease. AIDS Res. Hum. Retroviruses 2000, 16, 1209–1213. [Google Scholar] [CrossRef]
- Mammano, F.; Petit, C.; Clavel, F. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: Phenotypic analysis of protease and gag coevolution in protease inhibitor-treated patients. J. Virol. 1998, 72, 7632–7637. [Google Scholar] [CrossRef] [PubMed]
- Doyon, L.; Payant, C.; Brakier-Gingras, L.; Lamarre, D. Novel Gag-Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 1998, 72, 6146–6150. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; Parkin, N.; Tisdale, M.; Furfine, E.; Petropoulos, C.; Snowden, B.W.; Kleim, J.P. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Feher, A.; Weber, I.T.; Bagossi, P.; Baross, P.; Mahalingam, B.; Louis, J.M.; Copeland, T.D.; Yorshin, I.Y.; Harrison, R.W.; Tozser, J. Effect of sequence polymorphism and drug resistance on two HIV-1 Gag processing sites. J. Biochem. 2002, 269, 4114–4120. [Google Scholar] [CrossRef]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Krausslich, H.G.; Hance, A.J.; Clavel, F. Gag mutations strongly contribute to HIV-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; Heilek-Snyder, G.; Dulude, D.; Cammack, N.; Brakier-Gingras, L.; Konvalinka, J.; Parkin, N.; Krausslich, H.G.; Brun-Vezinet, F.; Boucher, C.A. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef]
- Kolli, M.; Lastere, S.; Schiffer, C.A. Co-evolution of nelfinavir-resistant HIV-1 protease and the p1-p6 substrate. Virology 2006, 347, 405–409. [Google Scholar] [CrossRef]
- Kolli, M.; Stawiski, E.; Chappey, C.; Schiffer, C.A. Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J. Virol. 2009, 83, 11027–11042. [Google Scholar] [CrossRef]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef]
- Martinez-Picado, J.; Savara, A.V.; Shi, L.; Sutton, L.; D'Aquila, R.T. Fitness of human immunodeficiency virus type 1 protease inhibitor-selected single mutants. Virology 2000, 275, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Savara, A.V.; Sutton, L.; D'Aquila, R.T. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 1999, 73, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Croteau, G.; Doyon, L.; Thibeault, D.; McKercher, G.; Pilote, L.; Lamarre, D. Impaired fitness of human immunodeficiency virus type 1 variants with high-level resistance to protease inhibitors. J. Virol. 1997, 71, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Bleiber, G.; Munoz, M.; Ciuffi, A.; Meylan, P.; Telenti, A. Individual contributions of mutant protease and reverse transcriptase to viral infectivity, replication, and protein maturation of antiretroviral drug-resistant human immunodeficiency virus type 1. J. Virol. 2001, 75, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.H.; Myers, R.E.; Snowden, B.W.; Tisdale, M.; Blair, E.D. HIV type 1 protease cleavage site mutations and viral fitness: Implications for drug susceptibility phenotyping assays. AIDS Res. Hum. Retroviruses 2000, 16, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Larrouy, L.; Chazallon, C.; Landman, R.; Capitant, C.; Peytavin, G.; Collin, G.; Charpentier, C.; Storto, A.; Pialoux, G.; Katlama, C.; Girard, P.M.; Yeni, P.; Aboulker, J.P.; Brun-Vezinet, F.; Descamps, D. Gag mutations can impact virological response to dual-boosted protease inhibitor combinations in antiretroviral-naive HIV-infected patients. Antimicrob. Agents Chemother. 2010, 54, 2910–2919. [Google Scholar] [CrossRef]
- Parry, C.M.; Kohli, A.; Boinett, C.J.; Towers, G.J.; McCormick, A.L.; Pillay, D. Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J. Virol. 2009, 83, 9094–9101. [Google Scholar] [CrossRef]
- Parkin, N.; Chappey, C.; Lam, E.; Petropoulos, C. Reduced susceptibility to protease inhibitors (PI) in the absence of primary PI resistance-associated mutations. Antivir. Ther. 2005, 10, S118. [Google Scholar]
- Clavel, F.; Mammano, F. Role of Gag in HIV Resistance to Protease Inhibitors. Viruses 2010, 2, 1411–1426. [Google Scholar] [CrossRef]
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; Learn, G.H.; Leitner, T.; McCutchan, F.; Osmanov, S.; Peeters, M.; Pieniazek, D.; Salminen, M.; Sharp, P.M.; Wolinsky, S.; Korber, B. HIV-1 nomenclature proposal. Science 2000, 288, 55–56. [Google Scholar] [CrossRef]
- HIV Sequence Database. Available online: http://www.hiv.lanl.gov/ (accessed on 20 October 2010).
- Velazquez-Campoy, A.; Vega, S.; Fleming, E.; Bacha, U.; Sayed, Y.; Dirr, H.W.; Freire, E. Protease inhibition in African subtypes of HIV-1. AIDS Rev. 2003, 5, 165–171. [Google Scholar] [PubMed]
- Bandaranayake, R.M.; Kolli, M.; King, N.M.; Nalivaika, E.; Heroux, A.; Kakizawa, J.; Sugiura, W.; Schiffer, C.A. The Effect of Clade Specific Sequence Polymorphisms on HIV-1 Protease Activity and Inhibitor Resistance Pathways. J. Virol. 2010, 84, 9995–10003. [Google Scholar] [CrossRef] [PubMed]
- Holguin, A.; Sune, C.; Hamy, F.; Soriano, V.; Klimkait, T. Natural polymorphisms in the protease gene modulate the replicative capacity of non-B HIV-1 variants in the absence of drug pressure. J. Clin. Virol. 2006, 36, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Paxinos, E.E.; Averbuch, D.; Maayan, S.; Parkin, N.T.; Engelhard, D.; Lorber, M.; Istomin, V.; Shaked, Y.; Mendelson, E.; Ram, D.; Petropoulos, C.J.; Schapiro, J.M. Mutation D30N is not preferentially selected by human immunodeficiency virus type 1 subtype C in the development of resistance to nelfinavir. Antimicrob. Agents Chemother. 2004, 48, 2159–2165. [Google Scholar] [CrossRef]
- Ariyoshi, K.; Matsuda, M.; Miura, H.; Tateishi, S.; Yamada, K.; Sugiura, W. Patterns of point mutations associated with antiretroviral drug treatment failure in CRF01_AE (subtype E) infection differ from subtype B infection. J. Acquir. Immune Defic. Syndr. 2003, 33, 336–342. [Google Scholar] [CrossRef]
- Lau, F.T.; Karplus, M. Molecular recognition in proteins. Simulation analysis of substrate binding by a tyrosyl-tRNA synthetase mutant. J. Mol. Biol. 1994, 236, 1049–1066. [Google Scholar] [CrossRef]
- Bash, P.A.; Singh, U.C.; Langridge, R.; Kollman, P.A. Free energy calculations by computer simulation. Science 1987, 287, 564–567. [Google Scholar] [CrossRef]
- Gao, J.; Kuczera, K.; Tidor, B.; Karplus, M. Hidden thermodynamics of mutant proteins: A molecular dynamics analysis. Science 1989, 244, 1069–1072. [Google Scholar] [CrossRef]
- Wong, C.F.; McCammon, J.A. Dynamics and Design of Enzymes and Inhibitors. J. Am. Chem. Soc. 1986, 108, 3830–3832. [Google Scholar] [CrossRef]
- Bash, P.A.; Singh, U.C.; Brown, F.K.; Langridge, R.; Kollman, P.A. Calculation of the relative change in binding free energy of a protein-inhibitor complex. Science 1987, 235, 574–576. [Google Scholar] [CrossRef]
- Kollman, P.A. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Purohit, R.; Sethumadhavan, R. Structural basis for the resilience of Darunavir (TMC114) resistance major flap mutations of HIV-1 protease. Interdiscip. Sci. 2009, 1, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discovery Des. 1999, 18, 113–135. [Google Scholar] [CrossRef]
- Swanson, J.M.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Stoica, I.; Sadiq, S.K.; Coveney, P.V. Rapid and accurate prediction of binding free energies for saquinavir-bound HIV-1 proteases. J. Am. Chem. Soc. 2008, 130, 2639–2648. [Google Scholar] [CrossRef]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 14937–14942. [Google Scholar] [CrossRef]
- Cai, Y.; Schiffer, C. Decomposing the Energetic Impact of Drug Resistant Mutations in HIV-1 Protease on Binding DRV. J. Chem. Theory Comput. 2010, 6, 1358–1368. [Google Scholar] [CrossRef]
- Singh, U.C.; Benkovic, S.J. A free-energy perturbation study of the binding of methotrexate to mutants of dihydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 9519–9523. [Google Scholar] [CrossRef]
- Blondel, A. Ensemble variance in free energy calculations by thermodynamic integration: Theory, optimal "Alchemical" path, and practical solutions. J. Comput. Chem. 2004, 25, 985–993. [Google Scholar] [CrossRef]
- Wlodawer, A.; Erickson, J.W. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 1993, 62, 543–585. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Archontis, G.; Simonson, T.; Karplus, M. Binding free energies and free energy components from molecular dynamics and Poisson-Boltzmann calculations. Application to amino acid recognition by aspartyl-tRNA synthetase. J. Mol. Biol. 2001, 306, 307–327. [Google Scholar] [CrossRef] [PubMed]
- Mardis, K.L.; Luo, R.; Gilson, M.K. Interpreting trends in the binding of cyclic ureas to HIV-1 protease. J. Mol. Biol. 2001, 309, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Altman, M.D.; Nalivaika, E.A.; Prabu-Jeyabalan, M.; Schiffer, C.A.; Tidor, B. Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease. Proteins 2008, 70, 678–694. [Google Scholar] [CrossRef]
- Huggins, D.J.; Altman, M.D.; Tidor, B. Evaluation of an inverse molecular design algorithm in a model binding site. Proteins 2009, 75, 168–186. [Google Scholar] [CrossRef]
- Sherman, W.; Tidor, B. Novel method for probing the specificity binding profile of ligands: Applications to HIV protease. Chem. Biol. Drug Des. 2008, 71, 387–407. [Google Scholar] [CrossRef]
- Chellappan, S.; Kairys, V.; Fernandes, M.X.; Schiffer, C.; Gilson, M.K. Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins 2007, 68, 561–567. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; King, N.M.; Nalivaika, E.A.; Heilek-Snyder, G.; Cammack, N.; Schiffer, C.A. Substrate envelope and drug resistance: Crystal structure of RO1 in complex with wild-type human immunodeficiency virus type 1 protease. Antimicrob. Agents Chemother. 2006, 50, 1518–1521. [Google Scholar] [CrossRef]
- Ali, A.; Reddy, G.S.K.K.; Cao, H.; Anjum, S.G.; Nalam, M.N.L.; Schiffer, C.A.; Rana, T.M. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl-oxazolidinone-5-carboxamides as novel P2 ligands. J. Med. Chem. 2006, 49, 7342–7356. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.; Reddy, G.S.K.K.; Ali, A.; Nalam, M.N.L.; Anjum, S.G.; Cao, H.; Kairys, V.; Fernandes, M.X.; Altman, M.D.; Tidor, B.; Rana, T.M.; Schiffer, C.A.; Gilson, M.K. Design of mutation-resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem. Biol. Drug Des. 2007, 69, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Kairys, V.; Gilson, M.K.; Lather, V.; Schiffer, C.A.; Fernandes, M.X. Toward the design of mutation-resistant enzyme inhibitors: Further evaluation of the substrate envelope hypothesis. Chem. Biol. Drug Des. 2009, 74, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Tuske, S.; Sarafianos, S.G.; Clark, A.D., Jr.; Ding, J.; Naeger, L.K.; White, K.L.; Miller, M.D.; Gibbs, C.S.; Boyer, P.L.; Clark, P.; Wang, G.; Gaffney, B.L.; Jones, R.A.; Jerina, D.M.; Hughes, S.H.; Arnold, E. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004, 11, 469–474. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Ki (nM) | Antiviral EC50 (nM) | |||
| WT/Q7K | L10I, G48V, I54V, L63P, V82A | D30N, L63P, N88D | L10I, L63P, A71V, G73S, I84V, L90M | ||
| Saquinavir | 0.065 | 90 | 1.0 | 78 | 26 |
| Indinavir | 0.18 | 34 | 0.73 | 21 | 40 |
| Ritonavir | 0.055 | 3.0 | 0.46 | 2.8 | 65 |
| Nelfinavir | 0.28 | 15 | 3.5 | 19 | 71 |
| Amprenavir | 0.10 | 0.15 | 0.21 | 1.40 | 44 |
| Lopinavir | 0.005 | 6.1 | 0.04 | 0.90 | 10 |
| Atazanavir | 0.046 | 0.33 | 0.009 | 0.49 | 15 |
| Tipranavir | 0.088 | 0.014 | 0.001 | 0.032 | 500 |
| Darunavir | 0.008 | 0.005 | 0.041 | 0.025 | 1 |
| Positions | Wild-type Amino Acid | Most Frequent Mutations | Polymorphic/Non-polymorphic |
|---|---|---|---|
| 10 | L | FI | (L10I) Polymorphic (L10F) Non-polymorphic |
| 11 | V | L | Non-polymorphic |
| 20 | K | T | Non-polymorphic |
| 33 | L | F | Non-polymorphic |
| 35 | E | GN | Non-polymorphic |
| 43 | K | T | Non-polymorphic |
| 46 | M | IL | Non-polymorphic |
| 54 | I | ALMSTV | Non-polymorphic |
| 58 | Q | E | Non-polymorphic |
| 73 | G | CST | Non-polymorphic |
| 74 | T | PS | Non-polymorphic |
| 76 | L | V | Non-polymorphic |
| 88 | N | DS | Non-polymorphic |
| 89 | L | V | Non-polymorphic |
| 90 | L | M | Non-polymorphic |
| Position | 10 | 12 | 13 | 14 | 15 | 20 | 35 | 36 | 41 | 57 | 61 | 69 | 82 | 89 | 93 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade B | L | T | I | K | I | K | E | M | R | R | Q | H | V | L | I |
| Resistance Associated Mutations in clade B | I | V | I/R | D | I | A | M | L | |||||||
| Clade A1/A2 | I | V | R | D | I | K | K | K | M | ||||||
| Clade C | S | V | I/V | K/N | K | M | L | ||||||||
| Clade D | V | V | I | K | |||||||||||
| Clade F1/F2 | V/I | S | V | R | D | I/V | K | K | N | M | |||||
| Clade G | I | V | R | I | D | I | K | K | I | M | |||||
| Clade H | V | R | I | K | I | ||||||||||
| Clade J | V | R | R | I | K | E | M | ||||||||
| Clade K | I | V | R | I | K | M | |||||||||
| CRF01_AE | V | R | D | I | K | K | M | ||||||||
| CRF02_AG | V/I | V | R | I | I | K | K | M |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.L.; Nalivaika, E.A.; Özen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509-2535. https://doi.org/10.3390/v2112509
Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MNL, Nalivaika EA, Özen A, et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses. 2010; 2(11):2509-2535. https://doi.org/10.3390/v2112509
Chicago/Turabian StyleAli, Akbar, Rajintha M. Bandaranayake, Yufeng Cai, Nancy M. King, Madhavi Kolli, Seema Mittal, Jennifer F. Murzycki, Madhavi N.L. Nalam, Ellen A. Nalivaika, Ayşegül Özen, and et al. 2010. "Molecular Basis for Drug Resistance in HIV-1 Protease" Viruses 2, no. 11: 2509-2535. https://doi.org/10.3390/v2112509
APA StyleAli, A., Bandaranayake, R. M., Cai, Y., King, N. M., Kolli, M., Mittal, S., Murzycki, J. F., Nalam, M. N. L., Nalivaika, E. A., Özen, A., Prabu-Jeyabalan, M. M., Thayer, K., & Schiffer, C. A. (2010). Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses, 2(11), 2509-2535. https://doi.org/10.3390/v2112509