Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C



Abstract
:1. Introduction
2. Viral Replication and Persistence
2.1. HIV
2.2. HBV
2.3. HCV
3. Virus Evolution in Individuals
3.1. HIV
3.2. HBV
3.3. HCV
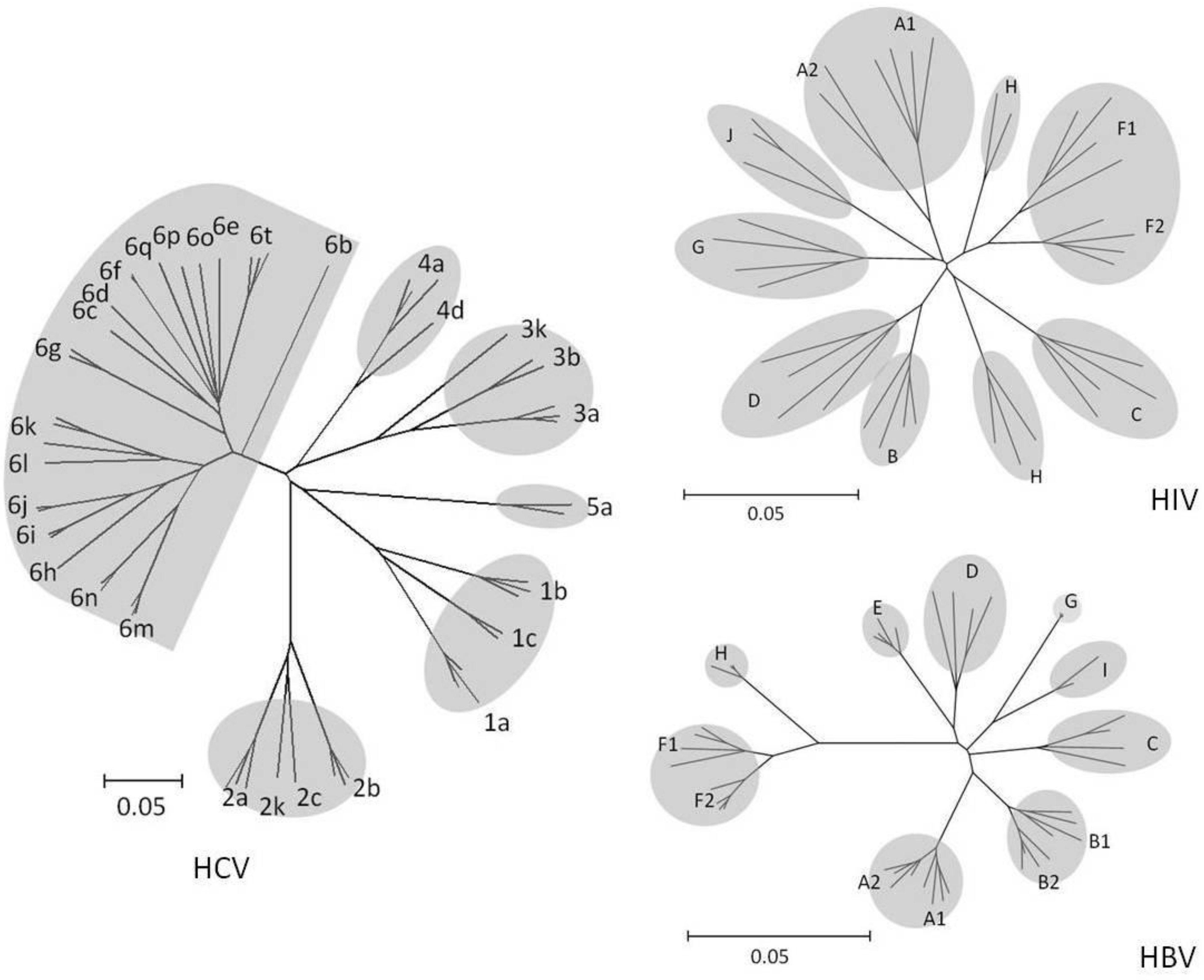
4. Virus Evolution in Populations
4.1. HIV
4.2. HBV
4.3. HCV
5. HIV Drug Resistance
5.1. Nucleoside/Nucleotide RT Inhibitors (NRTIs)
5.2. Nonnucleoside RT Inhibitor Resistance (NNRTIs)
5.3. Protease Inhibitors (PIs)
5.4. Integrase Inhibitors (INIs)
5.5. Fusion Inhibitors
5.6. CCR5 Inhibitors

{kind=link}
{kind=link}
| Drug Class | Mechanism of Resistance | Mutations | Drug Resistance Mutations |
|---|---|---|---|
| Nucleoside/Nucleotide RT inhibitors (NRTIs): Abacavir (ABC) Didanosine (ddI) Emtricitabine (FTC) Lamivudine (3TC) Stavudine (d4T) Zidovudine (AZT) Tenofovir (TDF) | RT mutations that enhance discrimination between NRTIs and natural nucleosides | K65R, L74V, Y115F, Q151M, M184V | K65R causes high-level resistance to ddI, ABC, and TDF, intermediate resistance to 3TC and FTC, low-level resistance to d4T, and increased susceptibility to AZT. L74V decreases susceptibility to ddI and ABC. Y115F decreases susceptibility to ABC and TDF. Q151M causes high-level resistance to AZT, d4T, ddI, and ABC, and intermediate resistance to TDF, 3TC, and FTC. M184V causes high-level resistance to 3TC and FTC and low-level resistance to ABC and ddI. Reviewed in [177,178]. |
| RT mutations that promote ATP-dependent hydrolytic removal of chain-terminating nucleotide monophosphates (also known as thymidine analog mutations or TAMs). | M41L, D67N, K70R, L210W, T215F/Y, K219Q/E T69S_SS | M41L, D67N, K70R, L210W, T215FY, and K219QE develop in viruses from patients receiving AZT and d4T. The accumulation of several TAMs causes cross-resistance to each of the other NRTIs except 3TC and FTC. T69S_SS is an uncommon amino acid insertion that confers resistance to each of the NRTIs when it occurs in combination with multiple TAMs. Reviewed in [177,178]. | |
| Non-nucleoside RT inhibitors (NNRTIs): Efavirenz (EFV) Etravirine (ETR) Nevirapine (NVP) | Mutations in the HIV-1 RT NNRTI-binding pocket | L100I, K101E/P, K103N, V106A/M Y181C/I/V, Y188L G190A/S, M230L | These mutations cause high-level resistance to NVP and intermediate or high-level resistance to EFV. With the exception of K103N, V106A/M, and Y188L, each mutation is also associated with decreased ETR susceptibility. Reviewed in [136,141,177]. |
| Protease inhibitors (PIs): Atazanavir (ATV) Darunavir (DRV) Fosamprenavir (FPV) Indinavir (IDV) Lopinavir/r (LPV/r) Nelfinavir (NFV) Saquinavir (SQV) Tipranavir (TPV) | Protease mutations interfere with inhibitor binding or compensate for the decreased replication associated with other mutations. | D30N, V32I, V47V/A, G48V, I50V/L, I54M/L/V/A/T, L76V, V82A/T/F/S/L, I84V/A, N88S, L90M | Positions 30, 32, 47, 48, 50, 82, and 84 are in the substrate cleft. Position 54 is in the flap and directly interacts with PIs as they enter the substrate cleft. The mutations at positions 76, 88, and 90 influence the shape of the substrate cleft indirectly. Reviewed in [142,177]. |
| These mutations are primarily compensatory | L10I/V/F, L24I, L33F, M46I/L F53L, A71V/T/I/L, Q58E, G73S/T/C/A, T74P, N83D, N88D, L89V | L10I/V, L33F, M46I/L, and A71V/T are minimally polymorphic occurring in 0.5% to 5% of viruses from untreated persons depending on the subtype. Reviewed in [142,177]. | |
| Integrase inhibitors (INIs): Raltegravir (RAL) In Phase III trials: Elvitegravir (EVG) S/GSK1349572 (572) | Mutations in residues surrounding the IN active site. | Q148H/R/K ± G140SA, N155H ± E92Q, Y143C/R, T66I/A/K, S147G | Q148H/R/K ± G140SA cause high-level RAL and EVG resistance and intermediate 572 resistance. N155H + E92Q causes high-level RAL and EVG resistance. Y143C/R + T97A causes high-level RAL resistance. T66I and S147G are selected in patients receiving EVG and decrease EVG susceptibility but do not appear to cause RAL cross-resistance. Reviewed in [179]. |
| Fusion inhibitors: Enfuvirtide (ENF) | Mutations in the first heptad repeat region (HR1) of the gp41 transmembrane protein interfere with the association of HR1 and HR2 required for virus cell fusion. | G36D/E/V/S, I37V, V38E/A/M/G, Q48H, N42T, N43D/K/S, L44M, L45M | G36D/E, V38E/A, Q40H, and N43D each reduce ENF susceptibility >10-fold [168,169]. Two mutations are usually sufficient to cause high-level ENF resistance. |
| CCR5 inhibitors: Maraviroc (MVC) | Virological failure and resistance is usually caused by expansion of pre-existing CXCR4-tropic variants that were not detected at the start of therapy. In vitro, and occasionally, in vivo resistance is caused by gp120 mutations that facilitate binding to an inhibitor bound CCR5 molecule. | Positively charged residues at positions 11 and 25 of the V3 loop of gp120 and many other combinations of mutations primarily but not exclusively within the V3 loop are associated with CXCR4 tropism [180]. No consistent pattern of gp120 mutations has been identified to be associated with virus binding to an inhibitor-bound CCR5 receptor [174,175,176]. | |
6. HBV Drug Resistance
6.1. Interferon (IFN)
6.2. Nucleoside/Nucleotide RT Inhibitors (NRTIs)
| Antiviral Agents | Mechanism of resistance | Mutations | Drug Resistance |
|---|---|---|---|
| Interferon | Unknown | Unknown | Unknown |
| Lamivudine (3TC) Telbivudine (LdT) Emtricitabine (FTC)* Entecavir (ETV) Adefovir (ADV) Tenofovir (TDF) | RT mutations that interfere with nucleotide triphosphate binding. Whether any of these mutations also facilitate primer unblocking is not known. | M204V/I ± L180M ± L80I, V173L | M204V/I ± L180M and less commonly L80I and V173L emerge during 3TC treatment and confer cross-resistance to LdT and FTC; and partial cross-resistance to ETV. M204V/I also emerge during LdT therapy. Reviewed in [215,223,226]. |
| N236T | Selected by ADV and causes partial cross-resistance to TDF. Reviewed in [215,223,226]. | ||
| A181V/T | Selected by ADV and less commonly 3TC. May causes partial cross-resistance to TDF but not ETV. Reviewed in [215,223,226]. | ||
| I169T, T184S/A/G, S202G/I, M250V | Selected by ETV particularly in viruses with pre-existing 3TC-resistance mutations. Reviewed in [215,223,226]. |
7. HCV Drug Resistance
7.1. Interferon (IFN) and Ribavirin
7.2. Protease Inhibitors (PIs)

7.3. Nucleoside (NI) and Non-nucleoside (NNI) Inhibitors
7.4. NS5A Inhibitors
7.5. Cyclophilin Inhibitors
| Antiviral Agents | Mechanism of resistance | Mutations | Drug Resistance |
|---|---|---|---|
| Interferon-α | Unknown | Genotype 1 isolates respond less well than genotype 2 or 3 viruses but the molecular basis is not known. | |
| Ribavirin | Unknown | Unknown | |
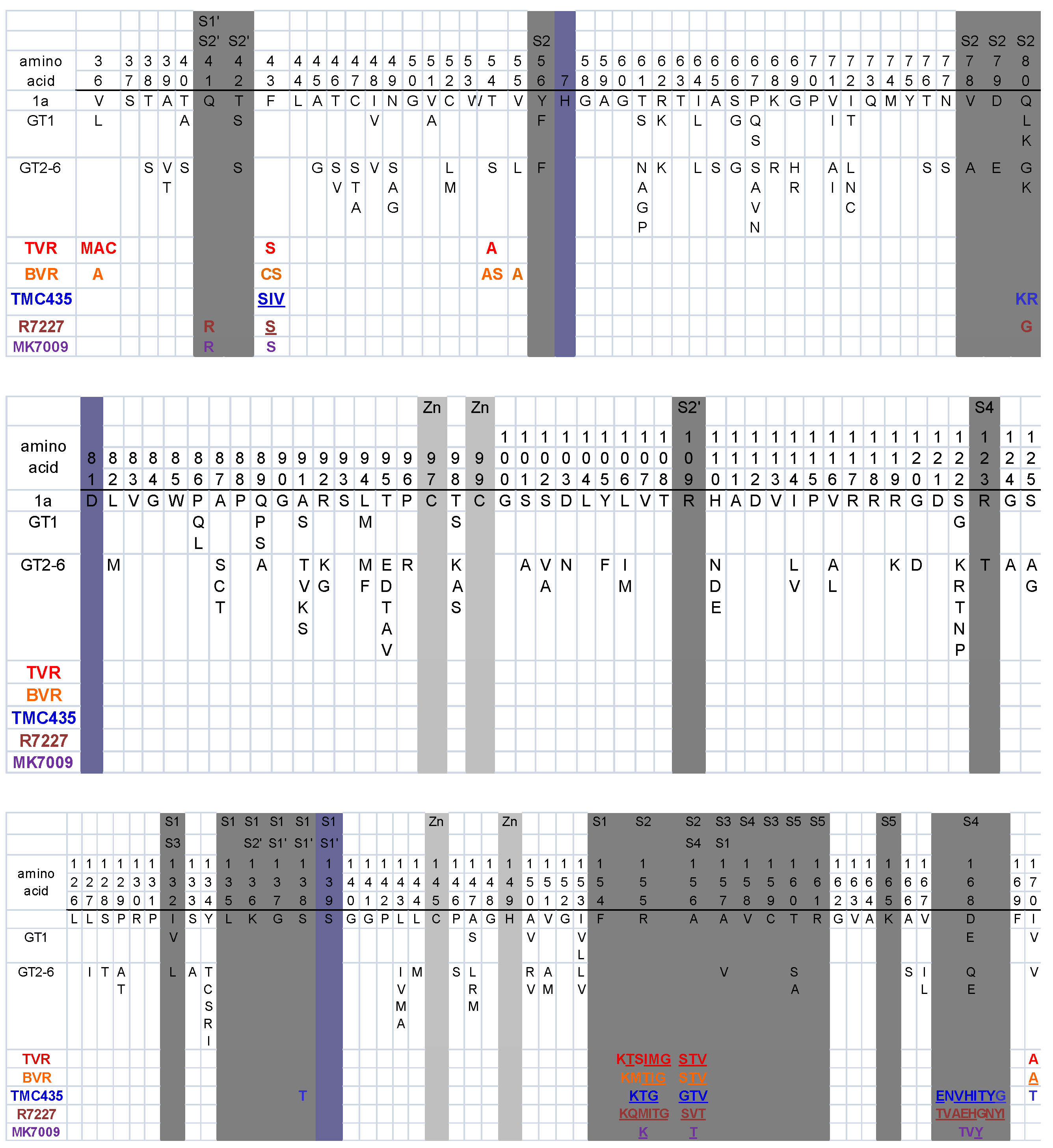
| PIs: Telaprevir (TVR) Boceprevir (BVR) TMC435 Danoprevir Vaniprevir | Mutations within or near the protease substrate cleft | V36A/M/C, Q41R, F43S/I/V, T54A/S, Q80K/R, R155K/T, A156S/V/T, D168A/E/I/N/T/V/Y, V170A/T | R155K/T and A156S/V/T decrease susceptibility to all PIs. V36A/M/C and T54A/S decrease susceptibility to the linear peptidomimetics TVR and BVR. V170A is selected by BVR but may cause cross-resistance to TVR. Q41R, F43S/I/V, and D168 mutations decrease susceptibility to TMC435, danoprevir, and vaniprevir. Q80K, a common polymorphism in genotype 1a, and V170T decrease TMC435 susceptibility about 5-fold.[233,263,268,271,272] |
| NIs: NM203 (withdrawn) R1626 (withdrawn) R7128 MK-0608 | Steric hindrance of nucleoside analog incorporation (S282T) | S282T | S282T in combination with compensatory mutations has been selected in vitro by 2’-C-methyl modified NIs including valopcitabine (NM203, an oral prodrug of the nucleoside analog 2’-C-methylcytidine) and R7128 (a pro-drug of PSI-6130). |
| S96T ± N142T | S96T ± N142T are selected in vitro by R1626 (a prodrug of R1479, 4’-azidocytidine). These mutations are far from the HCV polymerase active site. R1626 has been withdrawn from clinical development [288]. | ||
| NNIs | Decreased binding to NNI I pocket (upper thumb) | P495S/A/L, P496S/A, V499A | NNI site 1 is about 30Å from the active site [306]. A series of benzimidazole 5-carboxamide compounds bind to this site [292,307,308]. GS9190, BI207127, and MK3281 are site 1 NNIs in clinical development [68,233]. Mutations at positions 495, 496, and 497 reduce susceptibility to site 1 NNIs [68,233]. |
| Decreased binding to NNI site 2 (base of thumb) | L419V/M, M423T/V/I, I482L/V/T, V494A/I | NNI site 2 is a shallow hydrophobic pocket at the base of the thumb close to NNI site 1 and ~35Å from the active site. Compounds that bind to this site such as filibuvir, VC-759, and VCH-796 have selected the mutations L419M, M423T/V/I, I482L, and V194A [68,233,294,309]. | |
| Decreased binding to NNI site 3 (inner thumb / palm) | H95R, M414T, C451R, G554D, G558R, D559G | Benzothiadizine compounds that bind to this site have selected for M414T, C451R and G558R [310]. ANA598 is a site 3 NNI in Phase II trials [233]. Mutations associated with this drug have include M414T, G554D, and D559G [233]. M414T, which is polymorphic in genotypic 1 viruses, may play a role in resistance to both site 3 and site 4 NNIs. | |
| Decreased binding to NNI site 4 (palm) | C316N/Y, S365T, L392F, M414T, Y448H | C316Y is selected rapidly in vitro by HCV-796 [311], a site 4 NNI that is no longer in clinical development. Other mutations that have been selected by HCV-796 include C316N, S365T/A, L392F, and M414T [233,311]. ABT-333 is a site 4 NNI that has selected for C316N/Y and Y448H [233]. | |
| NS5A inhibitors: BMS-790052 | Unknown | M28T, Q30E/H/R, L31M/F/V, P32L, Y93C/H/N | In selection experiments with BMS-790052, M28T, Q30E/H/R, L31M/V, P32L, and Y93C/H/N have been selected in a genotype 1a replicon. L31F/V, P32L, and Y93H/N have been selected in vitro in a genotype 1b replicon. Two mutations are usually required for high-level resistance [302]. |
| Cyclophilin inhibitors: Debio 025 | Unknown | Unknown | |
8. Conclusions
Acknowledgements
References and Notes
- Han, Y.; Wind-Rotolo, M.; Yang, H.C.; Siliciano, J.D.; Siliciano, R.F. Experimental approaches to the study of HIV-1 latency. Nat. Rev. Microbiol. 2007, 5, 95–106. [Google Scholar] [CrossRef]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E.t.; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Yang, P.L.; Althage, A.; Chung, J.; Maier, H.; Wieland, S.; Isogawa, M.; Chisari, F.V. Immune effectors required for hepatitis B virus clearance. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 798–802. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection--natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef]
- Kenny-Walsh, E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. Irish Hepatology Research Group. N. Engl. J. Med. 1999, 340, 1228–1233. [Google Scholar] [CrossRef]
- Thomas, D.L.; Astemborski, J.; Rai, R.M.; Anania, F.A.; Schaeffer, M.; Galai, N.; Nolt, K.; Nelson, K.E.; Strathdee, S.A.; Johnson, L.; et al. The natural history of hepatitis C virus infection: Host, viral, and environmental factors. JAMA 2000, 284, 450–456. [Google Scholar] [CrossRef]
- Seeff, L.B.; Hollinger, F.B.; Alter, H.J.; Wright, E.C.; Cain, C.M.; Buskell, Z.J.; Ishak, K.G.; Iber, F.L.; Toro, D.; Samanta, A.; et al. Long-term mortality and morbidity of transfusion-associated non-A, non-B, and type C hepatitis: A National Heart, Lung, and Blood Institute collaborative study. Hepatology 2001, 33, 455–463. [Google Scholar] [CrossRef]
- Thompson, A.J.; Muir, A.J.; Sulkowski, M.S.; Ge, D.; Fellay, J.; Shianna, K.V.; Urban, T.; Afdhal, N.H.; Jacobson, I.M.; Esteban, R.; et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010, 139, 120–129.e118. [Google Scholar] [CrossRef]
- Keele, B.F.; Jones, J.H.; Terio, K.A.; Estes, J.D.; Rudicell, R.S.; Wilson, M.L.; Li, Y.; Learn, G.H.; Beasley, T.M.; Schumacher-Stankey, J.; et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 2009, 460, 515–519. [Google Scholar] [CrossRef]
- Bar, K.J.; Li, H.; Chamberland, A.; Tremblay, C.; Routy, J.P.; Grayson, T.; Sun, C.; Wang, S.; Learn, G.H.; Morgan, C.J.; et al. Wide variation in the multiplicity of HIV-1 infection among injection drug users. J. Virol. 2010, 84, 6241–6247. [Google Scholar] [CrossRef]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef]
- Davis, M.; Sagan, S.M.; Pezacki, J.P.; Evans, D.J.; Simmonds, P. Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J. Virol. 2008, 82, 11824–11836. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Worobey, M.; Woelk, C.H.; Holmes, E.C. Evidence for the non-quasispecies evolution of RNA viruses [corrected]. Mol. Biol. Evol. 2001, 18, 987–994. [Google Scholar] [CrossRef]
- Wilke, C.O. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 2005, 5, 44. [Google Scholar] [CrossRef]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Cheslock, S.R.; Zhang, W.H.; Hu, W.S.; Pathak, V.K. Retroviral mutation rates and reverse transcriptase fidelity. Front. Biosci. 2003, 8, d117–d134. [Google Scholar]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retrovirus. 1996, 12, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Coutsinos, D.; Invernizzi, C.F.; Xu, H.; Moisi, D.; Oliveira, M.; Brenner, B.G.; Wainberg, M.A. Template usage is responsible for the preferential acquisition of the K65R reverse transcriptase mutation in subtype C variants of human immunodeficiency virus type 1. J. Virol. 2009, 83, 2029–2033. [Google Scholar] [CrossRef]
- Hu, W.S.; Temin, H.M. Genetic consequences of packaging two RNA genomes in one retroviral particle: Pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4204–4209. [Google Scholar] [CrossRef]
- Ramirez, B.C.; Simon-Loriere, E.; Galetto, R.; Negroni, M. Implications of recombination for HIV diversity. Virus Res. 2008, 134, 64–73. [Google Scholar] [CrossRef]
- Neher, R.A.; Leitner, T. Recombination rate and selection strength in HIV intra-patient evolution. PLoS Comput. Biol. 2010, 6, e1000660. [Google Scholar] [CrossRef]
- Galli, A.; Kearney, M.; Nikolaitchik, O.A.; Yu, S.; Chin, M.P.; Maldarelli, F.; Coffin, J.M.; Pathak, V.K.; Hu, W.S. Patterns of Human Immunodeficiency Virus type 1 recombination ex vivo provide evidence for coadaptation of distant sites, resulting in purifying selection for intersubtype recombinants during replication. J. Virol. 2010, 84, 7651–7661. [Google Scholar] [CrossRef]
- Shankarappa, R.; Margolick, J.B.; Gange, S.J.; Rodrigo, A.G.; Upchurch, D.; Farzadegan, H.; Gupta, P.; Rinaldo, C.R.; Learn, G.H.; He, X.; et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 1999, 73, 10489–10502. [Google Scholar] [CrossRef]
- Piantadosi, A.; Chohan, B.; Panteleeff, D.; Baeten, J.M.; Mandaliya, K.; Ndinya-Achola, J.O.; Overbaugh, J. HIV-1 evolution in gag and env is highly correlated but exhibits different relationships with viral load and the immune response. AIDS 2009, 23, 579–587. [Google Scholar] [CrossRef]
- Kearney, M.; Maldarelli, F.; Shao, W.; Margolick, J.B.; Daar, E.S.; Mellors, J.W.; Rao, V.; Coffin, J.M.; Palmer, S. Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J. Virol. 2009, 83, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 2006, 80, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.L.; Kwon, P.; Nettles, R.E.; Han, Y.; Ray, S.C.; Siliciano, R.F. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 2005, 79, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J.; Rhee, S.Y.; Eriksson, N.; Liu, T.F.; Kiuchi, M.; Das, A.K.; Shafer, R.W. Sequence editing by Apolipoprotein B RNA-editing catalytic component [corrected] and epidemiological surveillance of transmitted HIV-1 drug resistance. AIDS 2008, 22, 717–725. [Google Scholar] [CrossRef]
- Jern, P.; Russell, R.A.; Pathak, V.K.; Coffin, J.M. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009, 5, e1000367. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 4398–4402. [Google Scholar] [CrossRef]
- Whalley, S.A.; Murray, J.M.; Brown, D.; Webster, G.J.; Emery, V.C.; Dusheiko, G.M.; Perelson, A.S. Kinetics of acute hepatitis B virus infection in humans. J. Exp. Med. 2001, 193, 847–854. [Google Scholar] [CrossRef]
- Ho, S.K.; Yam, W.C.; Leung, E.T.; Wong, L.P.; Leung, J.K.; Lai, K.N.; Chan, T.M. Rapid quantification of hepatitis B virus DNA by real-time PCR using fluorescent hybridization probes. J. Med. Microbiol. 2003, 52, 397–402. [Google Scholar] [CrossRef]
- Murray, J.M.; Purcell, R.H.; Wieland, S.F. The half-life of hepatitis B virions. Hepatology 2006, 44, 1117–1121. [Google Scholar] [CrossRef]
- Dandri, M.; Murray, J.M.; Lutgehetmann, M.; Volz, T.; Lohse, A.W.; Petersen, J. Virion half-life in chronic hepatitis B infection is strongly correlated with levels of viremia. Hepatology 2008, 48, 1079–1086. [Google Scholar] [CrossRef]
- Herz, A.V.; Bonhoeffer, S.; Anderson, R.M.; May, R.M.; Nowak, M.A. Viral dynamics in vivo: Limitations on estimates of intracellular delay and virus decay. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 7247–7251. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, C.; Horal, P.; Lindh, M. Long-term mutation rates in the hepatitis B virus genome. J. Gen. Virol. 2000, 81, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.A.; Holmes, E.C. A revised evolutionary history of hepatitis B virus (HBV). J. Mol. Evol. 2002, 54, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Osiowy, C.; Giles, E.; Tanaka, Y.; Mizokami, M.; Minuk, G.Y. Molecular evolution of hepatitis B virus over 25 years. J. Virol. 2006, 80, 10307–10314. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Khudyakov, Y. Coevolution and HBV drug resistance. Antivir. Ther. 2010, 15, 505–515. [Google Scholar] [CrossRef]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef]
- Gunther, S. Genetic variation in HBV infection: Genotypes and mutants. J. Clin. Virol. 2006, 36, S3–S11. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef]
- Mizokami, M.; Orito, E.; Ohba, K.; Ikeo, K.; Lau, J.Y.; Gojobori, T. Constrained evolution with respect to gene overlap of hepatitis B virus. J. Mol. Evol. 1997, 44, S83–S90. [Google Scholar] [CrossRef]
- Zaaijer, H.L.; van Hemert, F.J.; Koppelman, M.H.; Lukashov, V.V. Independent evolution of overlapping polymerase and surface protein genes of hepatitis B virus. J. Gen. Virol. 2007, 88, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Perelson, A.S.; Zoulim, F. Why are there different dynamics in the selection of drug resistance in HIV and hepatitis B and C viruses? J. Antimicrob. Chemother. 2008, 62, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.G.; Cheng, Y.; Guindon, S.; Seet, B.L.; Lee, L.Y.; Hu, P.; Wasser, S.; Peter, F.J.; Tan, T.; Goode, M.; et al. Viral quasi-species evolution during hepatitis Be antigen seroconversion. Gastroenterology 2007, 133, 951–958. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Q.; Yu, D.M.; Wan, M.B.; Zhang, X.X. Early changes of hepatitis B virus quasispecies during lamivudine treatment and the correlation with antiviral efficacy. J. Hepatol. 2009, 50, 895–905. [Google Scholar] [CrossRef]
- Margeridon-Thermet, S.; Shulman, N.S.; Ahmed, A.; Shahriar, R.; Liu, T.; Wang, C.; Holmes, S.P.; Babrzadeh, F.; Gharizadeh, B.; Hanczaruk, B.; et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J. Infect. Dis. 2009, 199, 1275–1285. [Google Scholar] [CrossRef]
- Pallier, C.; Rodriguez, C.; Brillet, R.; Nordmann, P.; Hezode, C.; Pawlotsky, J.M. Complex dynamics of hepatitis B Virus Res.istance to adefovir. Hepatology 2009, 49, 50–59. [Google Scholar] [CrossRef]
- Pallier, C.; Castera, L.; Soulier, A.; Hezode, C.; Nordmann, P.; Dhumeaux, D.; Pawlotsky, J.M. Dynamics of hepatitis B Virus Res.istance to lamivudine. J. Virol. 2006, 80, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.; Ishino, H.; Tsuge, M.; Fujimoto, Y.; Imamura, M.; Takahashi, S.; Chayama, K. G to A hypermutation of hepatitis B virus. Hepatology 2005, 41, 626–633. [Google Scholar] [CrossRef]
- Reuman, E.C.; Margeridon-Thermet, S.; Caudill, H.B.; Liu, T.; Borroto-Esoda, K.; Svarovskaia, E.S.; Holmes, S.; Shafer, R.W. A classification model for G-to-A hypermutation in hepatitis B virus ultra-deep pyrosequencing reads. Bioinformatics 2010, 26, 2929–2932. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Use and interpretation of virological tests for hepatitis C. Hepatology 2002, 36, S65–S73. [Google Scholar] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, B.; Bonhoeffer, S.; Binley, J.; Hurley, A.; Zhang, L.; Mittler, J.E.; Markowitz, M.; Moore, J.P.; Perelson, A.S.; Ho, D.D. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 1999, 354, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, E.; Neumann, A.U.; Schmidt, J.M.; Zeuzem, S. Hepatitis C virus kinetics. Antivir. Ther. 2000, 5, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Bichko, V.V.; Seeger, C. Effect of alpha interferon on the hepatitis C virus replicon. J. Virol. 2001, 75, 8516–8523. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 3392–3396. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Pathirana, S.; Davidson, F.; Lawlor, E.; Power, J.; Yap, P.L.; Simmonds, P. The origin of hepatitis C virus genotypes. J. Gen. Virol. 1997, 78 (Pt 2), 321–328. [Google Scholar] [CrossRef]
- Powdrill, M.; Bernatchez, J.; Gotte, M. Inhibitors of hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses 2010, 2, 2169–2195. [Google Scholar] [CrossRef]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus--15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef]
- Kalinina, O.; Norder, H.; Mukomolov, S.; Magnius, L.O. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J. Virol. 2002, 76, 4034–4043. [Google Scholar] [CrossRef]
- Colina, R.; Casane, D.; Vasquez, S.; Garcia-Aguirre, L.; Chunga, A.; Romero, H.; Khan, B.; Cristina, J. Evidence of intratypic recombination in natural populations of hepatitis C virus. J. Gen. Virol. 2004, 85, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Moreau, I.; Hegarty, S.; Levis, J.; Sheehy, P.; Crosbie, O.; Kenny-Walsh, E.; Fanning, L.J. Serendipitous identification of natural intergenotypic recombinants of hepatitis C in Ireland. Virol. J. 2006, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, M.U.; Cerino, A.; Meola, A.; Nicosia, A. Variability or conservation of hepatitis C virus hypervariable region 1? Implications for immune responses. J. Biosci. 2003, 28, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, I.; Pybus, O.G.; Holmes, E.C.; Klenerman, P. High-resolution phylogenetic analysis of hepatitis C virus adaptation and its relationship to disease progression. J. Virol. 2004, 78, 3447–3454. [Google Scholar] [CrossRef]
- Erickson, A.L.; Kimura, Y.; Igarashi, S.; Eichelberger, J.; Houghton, M.; Sidney, J.; McKinney, D.; Sette, A.; Hughes, A.L.; Walker, C.M. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 2001, 15, 883–895. [Google Scholar] [CrossRef]
- Soderholm, J.; Ahlen, G.; Kaul, A.; Frelin, L.; Alheim, M.; Barnfield, C.; Liljestrom, P.; Weiland, O.; Milich, D.R.; Bartenschlager, R.; et al. Relation between viral fitness and immune escape within the hepatitis C virus protease. Gut 2006, 55, 266–274. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Foy, E.M. Evasion of intracellular host defence by hepatitis C virus. Nature 2005, 436, 939–945. [Google Scholar] [CrossRef]
- Martell, M.; Esteban, J.I.; Quer, J.; Genesca, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gomez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar] [CrossRef]
- Manzin, A.; Solforosi, L.; Petrelli, E.; Macarri, G.; Tosone, G.; Piazza, M.; Clementi, M. Evolution of hypervariable region 1 of hepatitis C virus in primary infection. J. Virol. 1998, 72, 6271–6276. [Google Scholar] [CrossRef]
- Cristina, J.; del Pilar Moreno, M.; Moratorio, G. Hepatitis C virus genetic variability in patients undergoing antiviral therapy. Virus Res. 2007, 127, 185–194. [Google Scholar] [CrossRef]
- Fan, X.; Mao, Q.; Zhou, D.; Lu, Y.; Xing, J.; Xu, Y.; Ray, S.C.; Di Bisceglie, A.M. High diversity of hepatitis C viral quasispecies is associated with early virological response in patients undergoing antiviral therapy. Hepatology 2009, 50, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- McCutchan, F.E. Global epidemiology of HIV. J. Med. Virol. 2006, 78, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Parkin, N.T.; Schapiro, J.M. Antiretroviral drug resistance in non-subtype B HIV-1, HIV-2 and SIV. Antivir. Ther. 2004, 9, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.; Katzenstein, D.A.; Efron, B.; Carvalho, A.P.; Wynhoven, B.; Cane, P.; Clarke, J.; Sirivichayakul, S.; Soares, M.A.; Snoeck, J.; et al. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: Results of a global collaboration. PLoS Med. 2005, 2, e112. [Google Scholar] [CrossRef]
- Cane, P.A.; de Ruiter, A.; Rice, P.; Wiselka, M.; Fox, R.; Pillay, D. Resistance-associated mutations in the human immunodeficiency virus type 1 subtype c protease gene from treated and untreated patients in the United Kingdom. J. Clin. Microbiol. 2001, 39, 2652–2654. [Google Scholar] [CrossRef]
- Sugiura, W.; Matsuda, Z.; Yokomaku, Y.; Hertogs, K.; Larder, B.; Oishi, T.; Okano, A.; Shiino, T.; Tatsumi, M.; Matsuda, M.; et al. Interference between D30N and L90M in selection and development of protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2002, 46, 708–715. [Google Scholar] [CrossRef]
- Brenner, B.; Turner, D.; Oliveira, M.; Moisi, D.; Detorio, M.; Carobene, M.; Marlink, R.G.; Schapiro, J.; Roger, M.; Wainberg, M.A. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS 2003, 17, F1–F5. [Google Scholar] [CrossRef]
- Grossman, Z.; Istomin, V.; Averbuch, D.; Lorber, M.; Risenberg, K.; Levi, I.; Chowers, M.; Burke, M.; Bar Yaacov, N.; Schapiro, J.M. Genetic variation at NNRTI resistance-associated positions in patients infected with HIV-1 subtype C. AIDS 2004, 18, 909–915. [Google Scholar] [CrossRef]
- Grossman, Z.; Paxinos, E.E.; Averbuch, D.; Maayan, S.; Parkin, N.T.; Engelhard, D.; Lorber, M.; Istomin, V.; Shaked, Y.; Mendelson, E.; et al. Mutation D30N is not preferentially selected by human immunodeficiency virus type 1 subtype C in the development of resistance to nelfinavir. Antimicrob. Agents Chemother. 2004, 48, 2159–2165. [Google Scholar] [CrossRef]
- Camacho, R.; Godinho, A.; Gomes, P.; Abecasis, A.; Vandamme, A.-M.; Palma, C.; Carvalho, A.P.; Cabanas, J.; Goncalves, J. Different substitutions under drug pressure at protease codon 82 in HIV-1 subtype G compared to subtype B infected individuals including a novel I82M resistance mutations [abstract 138]. Antivir. Ther. 2005, 10, S151. [Google Scholar]
- Martinez-Cajas, J.L.; Pai, N.P.; Klein, M.B.; Wainberg, M.A. Differences in resistance mutations among HIV-1 non-subtype B infections: A systematic review of evidence (1996–2008). J. Int. AIDS Soc. 2009, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Doualla-Bell, F.; Avalos, A.; Brenner, B.; Gaolathe, T.; Mine, M.; Gaseitsiwe, S.; Oliveira, M.; Moisi, D.; Ndwapi, N.; Moffat, H.; et al. High prevalence of the K65R mutation in human immunodeficiency virus type 1 subtype C isolates from infected patients in Botswana treated with didanosine-based regimens. Antimicrob. Agents Chemother. 2006, 50, 4182–4185. [Google Scholar] [CrossRef] [PubMed]
- Hosseinipour, M.C.; van Oosterhout, J.J.; Weigel, R.; Phiri, S.; Kamwendo, D.; Parkin, N.; Fiscus, S.A.; Nelson, J.A.; Eron, J.J.; Kumwenda, J. The public health approach to identify antiretroviral therapy failure: High-level nucleoside reverse transcriptase inhibitor resistance among Malawians failing first-line antiretroviral therapy. AIDS 2009, 23, 1127–1134. [Google Scholar] [CrossRef]
- Invernizzi, C.F.; Coutsinos, D.; Oliveira, M.; Moisi, D.; Brenner, B.G.; Wainberg, M.A. Signature nucleotide polymorphisms at positions 64 and 65 in reverse transcriptase favor the selection of the K65R resistance mutation in HIV-1 subtype C. J. Infect. Dis. 2009, 200, 1202–1206. [Google Scholar] [CrossRef]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A new genotype of hepatitis B virus: Complete genome and phylogenetic relatedness. J. Gen. Virol. 2000, 81, 67–74. [Google Scholar] [CrossRef]
- Fung, S.K.; Lok, A.S. Hepatitis B virus genotypes: Do they play a role in the outcome of HBV infection? Hepatology 2004, 40, 790–792. [Google Scholar] [CrossRef]
- Liu, C.J.; Kao, J.H. Genetic variability of hepatitis B virus and response to antiviral therapy. Antivir. Ther. 2008, 13, 613–624. [Google Scholar] [CrossRef]
- Moucari, R.; Martinot-Peignoux, M.; Mackiewicz, V.; Boyer, N.; Ripault, M.P.; Castelnau, C.; Leclere, L.; Dauvergne, A.; Valla, D.; Vidaud, M.; et al. Influence of genotype on hepatitis B surface antigen kinetics in hepatitis B e antigen-negative patients treated with pegylated interferon-alpha2a. Antivir. Ther. 2009, 14, 1183–1188. [Google Scholar] [CrossRef]
- McMahon, B.J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol. Int. 2009, 3, 334–342. [Google Scholar] [CrossRef]
- Liaw, Y.; Brunetto, M.; Hadziyannis, S. The natural history of chronic HBV infection and geographical differences. Antivir. Ther. 2010, 15, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.N. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 2000, 13, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. The origin and evolution of hepatitis viruses in humans. J. Gen. Virol. 2001, 82, 693–712. [Google Scholar] [CrossRef]
- Legrand-Abravanel, F.; Colson, P.; Leguillou-Guillemette, H.; Alric, L.; Ravaux, I.; Lunel-Fabiani, F.; Bouviers-Alias, M.; Trimoulet, P.; Chaix, M.L.; Hezode, C.; et al. Influence of the HCV subtype on the virological response to pegylated interferon and ribavirin therapy. J. Med. Virol. 2009, 81, 2029–2035. [Google Scholar] [CrossRef]
- Ghany, M.G.; Strader, D.B.; Thomas, D.L.; Seeff, L.B. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2009, 49, 1335–1374. [Google Scholar] [CrossRef]
- Phillips, A.N.; Miller, V.; Sabin, C.; Cozzi Lepri, A.; Klauke, S.; Bickel, M.; Doerr, H.W.; Hill, A.; Staszewski, S. Durability of HIV-1 viral suppression over 3.3 years with multi-drug antiretroviral therapy in previously drug-naive individuals. AIDS 2001, 15, 2379–2384. [Google Scholar] [CrossRef]
- Bennett, D.; McCormick, L.; Kline, R.; Wheeler, W.; Hemmen, M.; Smith, A.; Zaidi, I.; Dondero, T. U.S. surveillance of HIV drug resistance at diagnosis using HIV diagnostic sera [Abstract 674]. In Proceeding of the 12th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 22–25 February 2005. [Google Scholar]
- Vercauteren, J.; Wensing, A.M.; van de Vijver, D.A.; Albert, J.; Balotta, C.; Hamouda, O.; Kucherer, C.; Struck, D.; Schmit, J.C.; Asjo, B.; et al. Transmission of drug-resistant HIV-1 is stabilizing in Europe. J. Infect. Dis. 2009, 200, 1503–1508. [Google Scholar] [CrossRef]
- Geretti, A.M. Epidemiology of antiretroviral drug resistance in drug-naive persons. Curr. Opin. Infect. Dis. 2007, 20, 22–32. [Google Scholar] [CrossRef]
- Chan, P.; Kantor, R. Transmitted drug resistance in nonsubtype B HIV-1 infection. HIV Therapy 2009, 3, 447–465. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; Schinazi, R.F.; So, A.G.; Scott, W.A. Differential removal of thymidine nucleotide analogues from blocked DNA chains by human immunodeficiency virus reverse transcriptase in the presence of physiological concentrations of 2'-deoxynucleoside triphosphates. Antimicrob. Agents Chemother. 2000, 44, 3465–3472. [Google Scholar] [CrossRef] [PubMed]
- Lennerstrand, J.; Stammers, D.K.; Larder, B.A. Biochemical mechanism of human immunodeficiency virus type 1 reverse transcriptase resistance to stavudine. Antimicrob. Agents Chemother. 2001, 45, 2144–2146. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, J.M.; Parkin, N.T.; Chappey, C.; Hellmann, N.S.; Petropoulos, C.J. Broad nucleoside reverse-transcriptase inhibitor cross-resistance in human immunodeficiency virus type 1 clinical isolates. J. Infect. Dis. 2003, 188, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Hughes, S.H.; Arnold, E. Taking aim at a moving target: Designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr. Opin. Struct. Biol. 2004, 14, 716–730. [Google Scholar] [CrossRef]
- Arion, D.; Sluis-Cremer, N.; Parniak, M.A. Mechanism by which phosphonoformic acid resistance mutations restore 3'- azido-3'-deoxythymidine (AZT) sensitivity to AZT-resistant HIV-1 reverse transcriptase. J. Biol. Chem. 2000, 275, 9251–9255. [Google Scholar] [CrossRef]
- Lanier, E.R.; Ait-Khaled, M.; Scott, J.; Stone, C.; Melby, T.; Sturge, G.; St Clair, M.; Steel, H.; Hetherington, S.; Pearce, G.; et al. Antiviral efficacy of abacavir in antiretroviral therapy-experienced adults harbouring HIV-1 with specific patterns of resistance to nucleoside reverse transcriptase inhibitors. Antivir. Ther. 2004, 9, 37–45. [Google Scholar] [CrossRef]
- Molina, J.M.; Marcelin, A.G.; Pavie, J.; Heripret, L.; De Boever, C.M.; Troccaz, M.; Leleu, G.; Calvez, V. Didanosine in HIV-1-infected patients experiencing failure of antiretroviral therapy: A randomized placebo-controlled trial. J. Infect. Dis. 2005, 191, 840–847. [Google Scholar] [CrossRef]
- Miller, M.D.; Margot, N.; Lu, B.; Zhong, L.; Chen, S.S.; Cheng, A.; Wulfsohn, M. Genotypic and phenotypic predictors of the magnitude of response to tenofovir disoproxil fumarate treatment in antiretroviral-experienced patients. J. Infect. Dis. 2004, 189, 837–846. [Google Scholar] [CrossRef]
- Parikh, U.M.; Koontz, D.L.; Chu, C.K.; Schinazi, R.F.; Mellors, J.W. In vitro activity of structurally diverse nucleoside analogs against human immunodeficiency virus type 1 with the K65R mutation in reverse transcriptase. Antimicrob. Agents Chemother. 2005, 49, 1139–1144. [Google Scholar] [CrossRef]
- Gallant, J.E.; DeJesus, E.; Arribas, J.R.; Pozniak, A.L.; Gazzard, B.; Campo, R.E.; Lu, B.; McColl, D.; Chuck, S.; Enejosa, J.; et al. Tenofovir DF, emtricitabine, and efavirenz vs. zidovudine, lamivudine, and efavirenz for HIV. N. Engl. J. Med. 2006, 354, 251–260. [Google Scholar] [CrossRef]
- Winters, M.A.; Coolley, K.L.; Girard, Y.A.; Levee, D.J.; Hamdan, H.; Shafer, R.W.; Katzenstein, D.A.; Merigan, T.C. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Invest. 1998, 102, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Masquelier, B.; Race, E.; Tamalet, C.; Descamps, D.; Izopet, J.; Buffet-Janvresse, C.; Ruffault, A.; Mohammed, A.S.; Cottalorda, J.; Schmuck, A.; et al. Genotypic and phenotypic resistance patterns of human immunodeficiency virus type 1 variants with insertions or deletions in the reverse transcriptase (RT): Multicenter study of patients treated with RT inhibitors. Antimicrob. Agents Chemother. 2001, 45, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Kozal, M.J.; Winters, M.A.; Iversen, A.K.; Katzenstein, D.A.; Ragni, M.V.; Meyer, W.A.; Gupta, P.; Rasheed, S.; Coombs, R.; et al. Combination therapy with zidovudine and didanosine selects for drug- resistant human immunodeficiency virus type 1 strains with unique patterns of pol gene mutations. J. Infect. Dis. 1994, 169, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, T.; Kavlick, M.F.; Ueno, T.; Gao, W.Y.; Kojima, E.; Alcaide, M.L.; Chokekijchai, S.; Roy, B.M.; Arnold, E.; Yarchoan, R.; et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2398–2402. [Google Scholar] [CrossRef] [PubMed]
- Ehteshami, M.; Gotte, M. Effects of mutations in the connection and RNase H domains of HIV-1 reverse transcriptase on drug susceptibility. AIDS Rev. 2008, 10, 224–235. [Google Scholar]
- Shahriar, R.; Rhee, S.Y.; Liu, T.F.; Fessel, W.J.; Scarsella, A.; Towner, W.; Holmes, S.P.; Zolopa, A.R.; Shafer, R.W. Nonpolymorphic human immunodeficiency virus type 1 protease and reverse transcriptase treatment-selected mutations. Antimicrob. Agents Chemother. 2009, 53, 4869–4878. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fransen, S.; Paxinos, E.E.; Stawiski, E.; Huang, W.; Petropoulos, C.J. Combinations of mutations in the connection domain of human immunodeficiency virus type 1 reverse transcriptase: Assessing the impact on nucleoside and nonnucleoside reverse transcriptase inhibitor resistance. Antimicrob. Agents Chemother. 2010, 54, 1973–1980. [Google Scholar] [CrossRef]
- Acosta-Hoyos, A.J.; Scott, W.A. The Role of Nucleotide Excision by Reverse Transcriptase in HIV Drug Resistance. Viruses 2010, 2, 372–394. [Google Scholar] [CrossRef]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Palmer, S.; Maldarelli, F.; Fivash, M.J., Jr.; Coffin, J.M.; Pathak, V.K. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3'-azido-3'-deoxythymidine resistance. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 317–322. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Pathak, V.K. The “connection” between HIV drug resistance and RNase H. Viruses 2010, 2, 1476–1503. [Google Scholar] [CrossRef]
- Parkin, N.T.; Hellmann, N.S.; Whitcomb, J.M.; Kiss, L.; Chappey, C.; Petropoulos, C.J. Natural variation of drug susceptibility in wild-type HIV-1. Antimicrob. Agents Chemother. 2004, 48, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B.; Becker-Pergola, G.; Guay, L.A.; Musoke, P.; Mracna, M.; Fowler, M.G.; Mofenson, L.M.; Mirochnick, M.; Mmiro, F.; Eshleman, S.H. Identification of the K103N resistance mutation in Ugandan women receiving nevirapine to prevent HIV-1 vertical transmission. AIDS 2000, 14, F111–F115. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, G.; Ngo-Giang-Huong, N.; Le Coeur, S.; Bowonwatanuwong, C.; Kantipong, P.; Leechanachai, P.; Ariyadej, S.; Leenasirimakul, P.; Hammer, S.; Lallemant, M. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine-based antiretroviral therapy. N. Engl. J. Med. 2004, 351, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoets, J.; Azijn, H.; Fransen, E.; De Baere, I.; Smeulders, L.; Jochmans, D.; Andries, K.; Pauwels, R.; de Bethune, M.P. TMC125 displays a high genetic barrier to the development of resistance: Evidence from in vitro selection experiments. J. Virol. 2005, 79, 12773–12782. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoets, J.; Tambuyzer, L.; Azijn, H.; Hoogstoel, A.; Nijs, S.; Peeters, M.; de Bethune, M.P.; De Smedt, G.; Woodfall, B.; Picchio, G. Resistance profile of etravirine: Combined analysis of baseline genotypic and phenotypic data from the randomized, controlled Phase III clinical studies. AIDS 2010, 24, 503–514. [Google Scholar] [CrossRef]
- Das, K.; Clark, A.D., Jr.; Lewi, P.J.; Heeres, J.; De Jonge, M.R.; Koymans, L.M.; Vinkers, H.M.; Daeyaert, F.; Ludovici, D.W.; Kukla, M.J.; et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar]
- Larder, B.A. Interactions between drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase. J. Gen. Virol. 1994, 75, 951–957. [Google Scholar] [CrossRef]
- Shulman, N.; Zolopa, A.R.; Passaro, D.; Shafer, R.W.; Huang, W.; Katzenstein, D.; Israelski, D.M.; Hellmann, N.; Petropoulos, C.; Whitcomb, J. Phenotypic hypersusceptibility to non-nucleoside reverse transcriptase inhibitors in treatment-experienced HIV-infected patients: Impact on virological response to efavirenz-based therapy. AIDS 2001, 15, 1125–1132. [Google Scholar] [CrossRef]
- Whitcomb, J.M.; Huang, W.; Limoli, K.; Paxinos, E.; Wrin, T.; Skowron, G.; Deeks, S.G.; Bates, M.; Hellmann, N.S.; Petropoulos, C.J. Hypersusceptibility to non-nucleoside reverse transcriptase inhibitors in HIV-1: Clinical, phenotypic and genotypic correlates. AIDS 2002, 16, F41–F47. [Google Scholar] [CrossRef]
- Vermeiren, H.; Van Craenenbroeck, E.; Alen, P.; Bacheler, L.; Picchio, G.; Lecocq, P. Prediction of HIV-1 drug susceptibility phenotype from the viral genotype using linear regression modeling. J. Virol. Methods 2007, 145, 47–55. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia, P.; Ruane, P.; Hellinger, J.; Shirvani, V.; Zolopa, A.; et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. [Google Scholar] [CrossRef]
- Erickson, J.W.; Gulnik, S.V.; Markowitz, M. Protease inhibitors: Resistance, cross-resistance, fitness and the choice of initial and salvage therapies. AIDS 1999, 13, S189–S204. [Google Scholar] [PubMed]
- Foulkes-Murzycki, J.E.; Scott, W.R.; Schiffer, C.A. Hydrophobic sliding: A possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 2007, 15, 225–233. [Google Scholar] [CrossRef]
- Cote, H.C.; Brumme, Z.L.; Harrigan, P.R. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 2001, 75, 589–594. [Google Scholar] [CrossRef]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; et al. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Kolli, M.; Lastere, S.; Schiffer, C.A. Co-evolution of nelfinavir-resistant HIV-1 protease and the p1-p6 substrate. Virology 2006, 347, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; et al. A Novel Substrate-Based HIV-1 Protease Inhibitor Drug Resistance Mechanism. PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Isaacson, J.D.; King, M.S.; Brun, S.C.; Xu, Y.; Real, K.; Bernstein, B.M.; Japour, A.J.; Sun, E.; Rode, R.A. Identification of genotypic changes in human immunodeficiency virus protease that correlate with reduced susceptibility to the protease inhibitor lopinavir among viral isolates from protease inhibitor-experienced patients. J. Virol. 2001, 75, 7462–7469. [Google Scholar] [CrossRef]
- De Meyer, S.; Azijn, H.; Surleraux, D.; Jochmans, D.; Tahri, A.; Pauwels, R.; Wigerinck, P.; de Bethune, M.P. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 2005, 49, 2314–2321. [Google Scholar] [CrossRef]
- Bierman, W.F.; van Agtmael, M.A.; Nijhuis, M.; Danner, S.A.; Boucher, C.A. HIV monotherapy with ritonavir-boosted protease inhibitors: A systematic review. AIDS 2009, 23, 279–291. [Google Scholar] [CrossRef]
- Arribas, J.R.; Horban, A.; Gerstoft, J.; Fatkenheuer, G.; Nelson, M.; Clumeck, N.; Pulido, F.; Hill, A.; van Delft, Y.; Stark, T.; et al. The MONET trial: Darunavir/ritonavir with or without nucleoside analogues, for patients with HIV RNA below 50 copies/mL. AIDS 2010, 24, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Katlama, C.; Valantin, M.A.; Algarte-Genin, M.; Duvivier, C.; Lambert-Niclot, S.; Girard, P.M.; Molina, J.M.; Hoen, B.; Pakianather, S.; Peytavin, G.; et al. Efficacy of darunavir/ritonavir maintenance monotherapy in patients with HIV-1 viral suppression: A randomized open-label, noninferiority trial, MONOI-ANRS 136. AIDS 2010, 24, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit HIV- 1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef]
- Johnson, A.A.; Santos, W.; Pais, G.C.; Marchand, C.; Amin, R.; Burke, T.R., Jr.; Verdine, G.; Pommier, Y. Integration requires a specific interaction of the donor DNA terminal 5'-cytosine with glutamine 148 of the HIV-1 integrase flexible loop. J. Biol. Chem. 2006, 281, 461–467. [Google Scholar] [CrossRef]
- McColl, D.J.; Chen, X. Strand transfer inhibitors of HIV-1 integrase: Bringing IN a new era of antiretroviral therapy. Antivir. Res. 2010, 85, 101–118. [Google Scholar] [CrossRef]
- Mouscadet, J.F.; Delelis, O.; Marcelin, A.G.; Tchertanov, L. Resistance to HIV-1 integrase inhibitors: A structural perspective. Drug Resist. Updat. 2010, 13, 139–150. [Google Scholar] [CrossRef] [PubMed]
- McColl, D.; Fransen, S.; Gupta, S.; Parking, N.; Margot, N.; Chuck, S.; Cheng, A.; Miller, M. Resistance and cross-resistance to first generation integrase inhibitors: Insights from a phase II study of elvitegravir (GS-9137) [abstract 9]. Antivir. Ther. 2007, 12, S11. [Google Scholar]
- Shimura, K.; Kodama, E.; Sakagami, Y.; Matsuzaki, Y.; Watanabe, W.; Yamataka, K.; Watanabe, Y.; Ohata, Y.; Doi, S.; Sato, M.; et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nakahara, K.; Seki, T.; Miki, S.; Kawauchi, S.; Suyama, A.; Wakasa-Morimoto, C.; Kodama, M.; Endoh, T.; Oosugi, E.; et al. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antivir. Res. 2008, 80, 213–222. [Google Scholar] [CrossRef]
- Goethals, O.; Clayton, R.; Van Ginderen, M.; Vereycken, I.; Wagemans, E.; Geluykens, P.; Dockx, K.; Strijbos, R.; Smits, V.; Vos, A.; et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008, 82, 10366–10374. [Google Scholar] [CrossRef]
- Goethals, O.; Vos, A.; Van Ginderen, M.; Geluykens, P.; Smits, V.; Schols, D.; Hertogs, K.; Clayton, R. Primary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profiles. Virology 2010, 402, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Gupta, S.; Danovich, R.; Hazuda, D.; Miller, M.; Witmer, M.; Petropoulos, C.J.; Huang, W. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 2009, 83, 11440–11446. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Malet, I.; Na, L.; Tchertanov, L.; Calvez, V.; Marcelin, A.G.; Subra, F.; Deprez, E.; Mouscadet, J.F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucl. Acids Res. 2009, 37, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Reuman, E.C.; Bachmann, M.H.; Varghese, V.; Fessel, W.J.; Shafer, R.W. Panel of prototypical raltegravir-resistant infectious molecular clones in a novel integrase-deleted cloning vector. Antimicrob. Agents Chemother. 2010, 54, 934–936. [Google Scholar] [CrossRef]
- Metifiot, M.; Maddali, K.; Naumova, A.; Zhang, X.; Marchand, C.; Pommier, Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 2010, 49, 3715–3722. [Google Scholar] [CrossRef]
- Kilby, J.M.; Eron, J.J. Novel therapies based on mechanisms of HIV-1 cell entry. N. Engl. J. Med. 2003, 348, 2228–2238. [Google Scholar] [CrossRef]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar] [CrossRef]
- Lu, J.; Deeks, S.G.; Hoh, R.; Beatty, G.; Kuritzkes, B.A.; Martin, J.N.; Kuritzkes, D.R. Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: Results of a clonal analysis. J. Acquir. Immune Defic. Syndr. 2006, 43, 60–64. [Google Scholar] [CrossRef]
- Hartley, O.; Klasse, P.; Sattentau, Q.; Moore, J.P. V3: HIV’s switch hitter. AIDS Res. Hum. Retrovirus. 2005, 21, 171–189. [Google Scholar] [CrossRef]
- Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 2007, 81, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using Virus Res.ervoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Kuritzkes, D.R. A piece de resistance: How HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV AIDS 2009, 4, 118–124. [Google Scholar] [CrossRef]
- Soriano, V.; Perno, C.F.; Kaiser, R.; Calvez, V.; Gatell, J.M.; di Perri, G.; Pillay, D.; Rockstroh, J.; Geretti, A.M. When and how to use maraviroc in HIV-infected patients. AIDS 2009, 23, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Doms, R.W. Entry inhibitors in the treatment of HIV-1 infection. Antivir. Res. 2010, 85, 91–100. [Google Scholar] [CrossRef]
- Shafer, R.W.; Schapiro, J.M. HIV-1 drug resistance mutations: An updated framework for the second decade of HAART. AIDS Rev. 2008, 10, 67–84. [Google Scholar]
- Singh, K.; Marchand, B.; Kirby, K.A.; Michailidis, E.; Sarafianos, S.G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606–638. [Google Scholar] [CrossRef]
- Metifiot, M.; Marchand, C.; Maddali, K.; Pommier, Y. Resistance to integrase inhibitors. Viruses 2010, 2, 1347–1366. [Google Scholar] [CrossRef]
- Low, A.J.; Swenson, L.C.; Harrigan, P.R. HIV coreceptor phenotyping in the clinical setting. AIDS Rev. 2008, 10, 143–151. [Google Scholar]
- Piccolo, P.; Lenci, I.; Demelia, L.; Bandiera, F.; Piras, M.R.; Antonucci, G.; Nosotti, L.; Mari, T.; De Santis, A.; Ponti, M.L.; et al. A randomized controlled trial of pegylated interferon-alpha2a plus adefovir dipivoxil for hepatitis B e antigen-negative chronic hepatitis B. Antivir. Ther. 2009, 14, 1165–1174. [Google Scholar] [CrossRef]
- Moucari, R.; Korevaar, A.; Lada, O.; Martinot-Peignoux, M.; Boyer, N.; Mackiewicz, V.; Dauvergne, A.; Cardoso, A.C.; Asselah, T.; Nicolas-Chanoine, M.H.; et al. High rates of HBsAg seroconversion in HBeAg-positive chronic hepatitis B patients responding to interferon: A long-term follow-up study. J. Hepatol. 2009, 50, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Takkenberg, B.; Zaaijer, H.; Weegink, C.; Terpstra, V.; Dijkgraaf, M.; Jansen, P.; Janssen, H.; Beld, M.; Reesinkg, H. Baseline HBsAg level predicts HBsAG loss in chronic hepatitis B patients treated with a combination of peginterferon alfa-2A and adefovir: An interim analysis. In Proceeding of the European Association Liver Disease 44th Annual Meeting, Copenhagen, Denmark, April 2009. [Google Scholar]
- Moucari, R.; Boyer, N.; Ripault, M.P.; Castelnau, C.; Mackiewicz, V.; Dauvergne, A.; Valla, D.; Vidaud, M.; Chanoine, M.H.; Marcellin, P. Sequential therapy with adefovir dipivoxil and pegylated Interferon Alfa-2a for HBeAg-negative patients. J. Viral. Hepat. 2010. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, A.; Tehan, B.G.; Chalmers, D.K. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir. Ther. 2004, 9, 149–160. [Google Scholar] [CrossRef]
- Stuyver, L.J.; Locarnini, S.A.; Lok, A.; Richman, D.D.; Carman, W.F.; Dienstag, J.L.; Schinazi, R.F. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology 2001, 33, 751–757. [Google Scholar] [CrossRef]
- Lai, C.L.; Dienstag, J.; Schiff, E.; Leung, N.W.; Atkins, M.; Hunt, C.; Brown, N.; Woessner, M.; Boehme, R.; Condreay, L. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin. Infect. Dis. 2003, 36, 687–696. [Google Scholar] [CrossRef]
- Marcellin, P.; Lau, G.K.; Bonino, F.; Farci, P.; Hadziyannis, S.; Jin, R.; Lu, Z.M.; Piratvisuth, T.; Germanidis, G.; Yurdaydin, C.; et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2004, 351, 1206–1217. [Google Scholar] [CrossRef]
- Lok, A.S.; Lai, C.L.; Leung, N.; Yao, G.B.; Cui, Z.Y.; Schiff, E.R.; Dienstag, J.L.; Heathcote, E.J.; Little, N.R.; Griffiths, D.A.; et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology 2003, 125, 1714–1722. [Google Scholar] [CrossRef]
- Tipples, G.A.; Ma, M.M.; Fischer, K.P.; Bain, V.G.; Kneteman, N.M.; Tyrrell, D.L. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo. Hepatology 1996, 24, 714–717. [Google Scholar] [CrossRef]
- Allen, M.I.; Deslauriers, M.; Andrews, C.W.; Tipples, G.A.; Walters, K.A.; Tyrrell, D.L.; Brown, N.; Condreay, L.D. Identification and characterization of mutations in hepatitis B Virus Res.istant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998, 27, 1670–1677. [Google Scholar] [CrossRef]
- Ono, S.K.; Kato, N.; Shiratori, Y.; Kato, J.; Goto, T.; Schinazi, R.F.; Carrilho, F.J.; Omata, M. The polymerase L528M mutation cooperates with nucleotide binding-site mutations, increasing hepatitis B virus replication and drug resistance. J. Clin. Invest. 2001, 107, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Delaney, W.E.t.; Yang, H.; Westland, C.E.; Das, K.; Arnold, E.; Gibbs, C.S.; Miller, M.D.; Xiong, S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003, 77, 11833–11841. [Google Scholar] [CrossRef] [PubMed]
- Warner, N.; Locarnini, S.; Kuiper, M.; Bartholomeusz, A.; Ayres, A.; Yuen, L.; Shaw, T. The L80I substitution in the reverse transcriptase domain of the hepatitis B virus polymerase is associated with lamivudine resistance and enhanced viral replication in vitro. Antimicrob. Agents Chemother. 2007, 51, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Liaw, Y.F.; Gane, E.; Leung, N.; Zeuzem, S.; Wang, Y.; Lai, C.L.; Heathcote, E.J.; Manns, M.; Bzowej, N.; Niu, J.; et al. 2-Year GLOBE trial results: Telbivudine Is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology 2009, 136, 486–495. [Google Scholar] [CrossRef]
- Tenney, D.J.; Levine, S.M.; Rose, R.E.; Walsh, A.W.; Weinheimer, S.P.; Discotto, L.; Plym, M.; Pokornowski, K.; Yu, C.F.; Angus, P.; et al. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob. Agents Chemother. 2004, 48, 3498–3507. [Google Scholar] [CrossRef]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Levine, S.M.; Pokornowski, K.A.; Walsh, A.W.; Fang, J.; Yu, C.F.; Zhang, S.; Mazzucco, C.E.; et al. Two-year assessment of entecavir resistance in Lamivudine-refractory hepatitis B virus patients reveals different clinical outcomes depending on the resistance substitutions present. Antimicrob. Agents Chemother. 2007, 51, 902–911. [Google Scholar] [CrossRef]
- Sherman, M.; Yurdaydin, C.; Simsek, H.; Silva, M.; Liaw, Y.F.; Rustgi, V.K.; Sette, H.; Tsai, N.; Tenney, D.J.; Vaughan, J.; et al. Entecavir therapy for lamivudine-refractory chronic hepatitis B: Improved virologic, biochemical, and serology outcomes through 96 weeks. Hepatology 2008, 48, 99–108. [Google Scholar] [CrossRef]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; et al. Long-term monitoring shows hepatitis B Virus Res.istance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef]
- Choe, W.H.; Hong, S.P.; Kim, B.K.; Ko, S.Y.; Jung, Y.K.; Kim, J.H.; Yeon, J.E.; Byun, K.S.; Kim, K.H.; Ji, S.I.; et al. Evolution of hepatitis B virus mutation during entecavir rescue therapy in patients with antiviral resistance to lamivudine and adefovir. Antivir. Ther. 2009, 14, 985–993. [Google Scholar] [CrossRef]
- Chang, T.T.; Gish, R.G.; Hadziyannis, S.J.; Cianciara, J.; Rizzetto, M.; Schiff, E.R.; Pastore, G.; Bacon, B.R.; Poynard, T.; Joshi, S.; et al. A dose-ranging study of the efficacy and tolerability of entecavir in Lamivudine-refractory chronic hepatitis B patients. Gastroenterology 2005, 129, 1198–1209. [Google Scholar] [CrossRef]
- Westland, C.E.; Yang, H.; Delaney, W.E.t.; Gibbs, C.S.; Miller, M.D.; Wulfsohn, M.; Fry, J.; Brosgart, C.L.; Xiong, S. Week 48 resistance surveillance in two phase 3 clinical studies of adefovir dipivoxil for chronic hepatitis B. Hepatology 2003, 38, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Tassopoulos, N.C.; Heathcote, E.J.; Chang, T.T.; Kitis, G.; Rizzetto, M.; Marcellin, P.; Lim, S.G.; Goodman, Z.; Ma, J.; et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2005, 352, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Tassopoulos, N.C.; Heathcote, E.J.; Chang, T.T.; Kitis, G.; Rizzetto, M.; Marcellin, P.; Lim, S.G.; Goodman, Z.; Ma, J.; et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology 2006, 131, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Angus, P.; Vaughan, R.; Xiong, S.; Yang, H.; Delaney, W.; Gibbs, C.; Brosgart, C.; Colledge, D.; Edwards, R.; Ayres, A.; et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology 2003, 125, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.K.; Chae, H.B.; Fontana, R.J.; Conjeevaram, H.; Marrero, J.; Oberhelman, K.; Hussain, M.; Lok, A.S. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J. Hepatol. 2006, 44, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Esoda, K.; Miller, M.D.; Arterburn, S. Pooled analysis of amino acid changes in the HBV polymerase in patients from four major adefovir dipivoxil clinical trials. J. Hepatol. 2007, 47, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Santantonio, T.; Fasano, M.; Durantel, S.; Barraud, L.; Heichen, M.; Guastadisegni, A.; Pastore, G.; Zoulim, F. Adefovir dipivoxil resistance patterns in patients with lamivudine-resistant chronic hepatitis B. Antivir. Ther. 2009, 14, 557–565. [Google Scholar] [CrossRef]
- Yang, H.; Westland, C.; Xiong, S.; Delaney, W.E.t. In vitro antiviral susceptibility of full-length clinical hepatitis B virus isolates cloned with a novel expression vector. Antivir. Res. 2004, 61, 27–36. [Google Scholar] [CrossRef]
- Peters, M.G.; Hann Hw, H.; Martin, P.; Heathcote, E.J.; Buggisch, P.; Rubin, R.; Bourliere, M.; Kowdley, K.; Trepo, C.; Gray Df, D.; et al. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 2004, 126, 91–101. [Google Scholar] [CrossRef]
- Westland, C.E.; Yang, H.; Delaney, W.E.t.; Wulfsohn, M.; Lama, N.; Gibbs, C.S.; Miller, M.D.; Fry, J.; Brosgart, C.L.; Schiff, E.R.; et al. Activity of adefovir dipivoxil against all patterns of lamivudine-resistant hepatitis B viruses in patients. J. Viral. Hepat. 2005, 12, 67–73. [Google Scholar] [CrossRef]
- Yang, H.; Qi, X.; Sabogal, A.; Miller, M.; Xiong, S.; Delaney, W.E.t. Cross-resistance testing of next-generation nucleoside and nucleotide analogues against lamivudine-resistant HBV. Antivir. Ther. 2005, 10, 625–633. [Google Scholar] [CrossRef]
- Rapti, I.; Dimou, E.; Mitsoula, P.; Hadziyannis, S.J. Adding-on versus switching-to adefovir therapy in lamivudine-resistant HBeAg-negative chronic hepatitis B. Hepatology 2007, 45, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Zoulim, F.; Locarnini, S.; Bartholomeusz, A.; Ghany, M.G.; Pawlotsky, J.M.; Liaw, Y.F.; Mizokami, M.; Kuiken, C. Antiviral drug-resistant HBV: Standardization of nomenclature and assays and recommendations for management. Hepatology 2007, 46, 254–265. [Google Scholar] [CrossRef]
- Keeffe, E.B.; Dieterich, D.T.; Pawlotsky, J.M.; Benhamou, Y. Chronic hepatitis B: Preventing, detecting, and managing viral resistance. Clin. Gastroenterol. Hepatol. 2008, 6, 268–274. [Google Scholar] [CrossRef]
- Lok, A.S.; McMahon, B.J. Chronic hepatitis B: Update 2009. Hepatology 2009, 50, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.T.; Chien, R.N.; Chu, C.M.; Liaw, Y.F. Clearance of the original hepatitis B virus YMDD-motif mutants with emergence of distinct lamivudine-resistant mutants during prolonged lamivudine therapy. Hepatology 2000, 31, 1318–1326. [Google Scholar] [CrossRef]
- Yatsuji, H.; Noguchi, C.; Hiraga, N.; Mori, N.; Tsuge, M.; Imamura, M.; Takahashi, S.; Iwao, E.; Fujimoto, Y.; Ochi, H.; et al. Emergence of a novel lamivudine-resistant hepatitis B virus variant with a substitution outside the YMDD motif. Antimicrob. Agents Chemother. 2006, 50, 3867–3874. [Google Scholar] [CrossRef]
- Gerolami, R.; Bourliere, M.; Colson, P.; Halfon, P.; Borentain, P.; Henry, M.; Botta, D.; Thibault, V.; Khiri, H.; Tamalet, C. Unusual selection of rtA181V HBV mutants cross-resistant to adefovir following prolonged lamivudine monotherapy: Report of two cases. Antivir. Ther. 2006, 11, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Villet, S.; Pichoud, C.; Billioud, G.; Barraud, L.; Durantel, S.; Trepo, C.; Zoulim, F. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J. Hepatol. 2008, 48, 747–755. [Google Scholar] [CrossRef]
- Warner, N.; Locarnini, S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology 2008, 48, 88–98. [Google Scholar] [CrossRef]
- Zoulim, F.; Locarnini, S. Hepatitis B Virus Res.istance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608.e1591–1592. [Google Scholar] [CrossRef]
- Tan, J.; Degertekin, B.; Wong, S.N.; Husain, M.; Oberhelman, K.; Lok, A.S. Tenofovir monotherapy is effective in hepatitis B patients with antiviral treatment failure to adefovir in the absence of adefovir-resistant mutations. J. Hepatol. 2008, 48, 391–398. [Google Scholar] [CrossRef] [PubMed]
- van Bommel, F.; de Man, R.A.; Wedemeyer, H.; Deterding, K.; Petersen, J.; Buggisch, P.; Erhardt, A.; Huppe, D.; Stein, K.; Trojan, J.; et al. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology 2010, 51, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Bartholomeusz, A.; Locarnini, S. HBV drug resistance: Mechanisms, detection and interpretation. J. Hepatol. 2006, 44, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, O.; Olotu, C.; Funk, A.; Zollner, B.; Helm, M.; Rockstroh, J.K.; Sirma, H. Selection and counterselection of the adefovir resistance mutation rtI233V during antiviral therapy. J. Clin. Microbiol. 2009. [Google Scholar] [CrossRef]
- Schildgen, O.; Sirma, H.; Funk, A.; Olotu, C.; Wend, U.C.; Hartmann, H.; Helm, M.; Rockstroh, J.K.; Willems, W.R.; Will, H.; et al. Variant of hepatitis B virus with primary resistance to adefovir. N. Engl. J. Med. 2006, 354, 1807–1812. [Google Scholar] [CrossRef]
- Curtis, M.; Zhu, Y.; Borroto-Esoda, K. Hepatitis B virus containing the I233V mutation in the polymerase reverse-transcriptase domain remains sensitive to inhibition by adefovir. J. Infect. Dis. 2007, 196, 1483–1486. [Google Scholar] [CrossRef]
- Sheldon, J.; Camino, N.; Rodes, B.; Bartholomeusz, A.; Kuiper, M.; Tacke, F.; Nunez, M.; Mauss, S.; Lutz, T.; Klausen, G.; et al. Selection of hepatitis B virus polymerase mutations in HIV-coinfected patients treated with tenofovir. Antivir. Ther. 2005, 10, 727–734. [Google Scholar] [CrossRef]
- Qi, X.; Xiong, S.; Yang, H.; Miller, M.; Delaney, W.E.t. In vitro susceptibility of adefovir-associated hepatitis B virus polymerase mutations to other antiviral agents. Antivir. Ther. 2007, 12, 355–362. [Google Scholar] [CrossRef]
- Burton, J.R., Jr.; Everson, G.T. HCV NS5B polymerase inhibitors. Clin. Liver Dis. 2009, 13, 453–465. [Google Scholar] [CrossRef]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Naggie, S.; Patel, K.; McHutchison, J. Hepatitis C virus directly acting antivirals: Current developments with NS3/4A HCV serine protease inhibitors. J. Antimicrob. Chemother. 2010, 65, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Villanueva, R.A.; Thomas, D.L.; Wakita, T.; Lemon, S.M. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2310–2315. [Google Scholar] [CrossRef]
- Kato, T.; Matsumura, T.; Heller, T.; Saito, S.; Sapp, R.K.; Murthy, K.; Wakita, T.; Liang, T.J. Production of infectious hepatitis C virus of various genotypes in cell cultures. J. Virol. 2007, 81, 4405–4411. [Google Scholar] [CrossRef]
- Pang, P.S.; Planet, P.J.; Glenn, J.S. The evolution of the major hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS One 2009, 4, e6579. [Google Scholar] [CrossRef]
- Asselah, T.; Estrabaud, E.; Bieche, I.; Lapalus, M.; De Muynck, S.; Vidaud, M.; Saadoun, D.; Soumelis, V.; Marcellin, P. Hepatitis C: Viral and host factors associated with non-response to pegylated interferon plus ribavirin. Liver Int. 2010, 30, 1259–1269. [Google Scholar] [CrossRef]
- Jaeckel, E.; Cornberg, M.; Wedemeyer, H.; Santantonio, T.; Mayer, J.; Zankel, M.; Pastore, G.; Dietrich, M.; Trautwein, C.; Manns, M.P. Treatment of acute hepatitis C with interferon alfa-2b. N. Engl. J. Med. 2001, 345, 1452–1457. [Google Scholar] [CrossRef]
- Gerlach, J.T.; Diepolder, H.M.; Zachoval, R.; Gruener, N.H.; Jung, M.C.; Ulsenheimer, A.; Schraut, W.W.; Schirren, C.A.; Waechtler, M.; Backmund, M.; et al. Acute hepatitis C: High rate of both spontaneous and treatment-induced viral clearance. Gastroenterology 2003, 125, 80–88. [Google Scholar] [CrossRef]
- Farci, P.; Strazzera, R.; Alter, H.J.; Farci, S.; Degioannis, D.; Coiana, A.; Peddis, G.; Usai, F.; Serra, G.; Chessa, L.; et al. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 3081–3086. [Google Scholar] [CrossRef] [PubMed]
- Morishima, C.; Polyak, S.J.; Ray, R.; Doherty, M.C.; Di Bisceglie, A.M.; Malet, P.F.; Bonkovsky, H.L.; Sullivan, D.G.; Gretch, D.R.; Rothman, A.L.; et al. Hepatitis C virus-specific immune responses and quasi-species variability at baseline are associated with nonresponse to antiviral therapy during advanced hepatitis C. J. Infect. Dis. 2006, 193, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Veillon, P.; Payan, C.; Le Guillou-Guillemette, H.; Gaudy, C.; Lunel, F. Quasispecies evolution in NS5A region of hepatitis C virus genotype 1b during interferon or combined interferon-ribavirin therapy. World J. Gastroenterol. 2007, 13, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Aurora, R.; Donlin, M.J.; Cannon, N.A.; Tavis, J.E. Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans. J. Clin. Invest. 2009, 119, 225–236. [Google Scholar] [CrossRef]
- Chary, A.; Winters, M.A.; Kottilil, S.; Murphy, A.A.; Polis, M.A.; Holodniy, M. Impact of Interferon-Ribavirin Treatment on Hepatitis C Virus (HCV) Protease Quasispecies Diversity in HIV- and HCV-Coinfected Patients. J. Infect. Dis. 2010, 202, 889–893. [Google Scholar] [CrossRef]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996, 334, 77–81. [Google Scholar] [CrossRef]
- Witherell, G.W.; Beineke, P. Statistical analysis of combined substitutions in nonstructural 5A region of hepatitis C virus and interferon response. J. Med. Virol. 2001, 63, 8–16. [Google Scholar] [CrossRef]
- Guo, J.T.; Sohn, J.A.; Zhu, Q.; Seeger, C. Mechanism of the interferon alpha response against hepatitis C virus replicons. Virology 2004, 325, 71–81. [Google Scholar] [CrossRef]
- Brillet, R.; Penin, F.; Hezode, C.; Chouteau, P.; Dhumeaux, D.; Pawlotsky, J.M. The nonstructural 5A protein of hepatitis C virus genotype 1b does not contain an interferon sensitivity-determining region. J. Infect. Dis. 2007, 195, 432–441. [Google Scholar] [CrossRef]
- Cannon, N.A.; Donlin, M.J.; Fan, X.; Aurora, R.; Tavis, J.E. Hepatitis C virus diversity and evolution in the full open-reading frame during antiviral therapy. PLoS One 2008, 3, e2123. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Torres-Puente, M.; Jimenez-Hernandez, N.; Bracho, M.A.; Garcia-Robles, I.; Wrobel, B.; Carnicer, F.; del Olmo, J.; Ortega, E.; Moya, A.; et al. Genetic variability of hepatitis C virus before and after combined therapy of interferon plus ribavirin. PLoS One 2008, 3, e3058. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.Y.; Tam, R.C.; Liang, T.J.; Hong, Z. Mechanism of action of ribavirin in the combination treatment of chronic HCV infection. Hepatology 2002, 35, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Shields, W.W.; Pockros, P.J. Ribavirin analogs. Clin. Liver Dis. 2009, 13, 419–427. [Google Scholar] [CrossRef]
- Maag, D.; Castro, C.; Hong, Z.; Cameron, C.E. Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 2001, 276, 46094–46098. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Chevaliez, S.; Brillet, R.; Lazaro, E.; Hezode, C.; Pawlotsky, J.M. Analysis of ribavirin mutagenicity in human hepatitis C virus infection. J. Virol. 2007, 81, 7732–7741. [Google Scholar] [CrossRef]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Kwong, A.D.; Kim, J.L.; Rao, G.; Lipovsek, D.; Raybuck, S.A. Hepatitis C virus NS3/4A protease. Antivir. Res. 1999, 41, 67–84. [Google Scholar]
- Landro, J.A.; Raybuck, S.A.; Luong, Y.P.; O’Malley, E.T.; Harbeson, S.L.; Morgenstern, K.A.; Rao, G.; Livingston, D.J. Mechanistic role of an NS4A peptide cofactor with the truncated NS3 protease of hepatitis C virus: Elucidation of the NS4A stimulatory effect via kinetic analysis and inhibitor mapping. Biochemistry 1997, 36, 9340–9348. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Kwong, A.D.; Picchio, G.R. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J. Antimicrob. Chemother. 2010, 65, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hezode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J.P.; Bourliere, M.; Gharakhanian, S.; Bengtsson, L.; et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009, 360, 1839–1850. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Manns, M.P.; Muir, A.J.; Terrault, N.A.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.J.; Zeuzem, S.; Reesink, H.W.; Garg, J.; et al. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 2010, 362, 1292–1303. [Google Scholar] [CrossRef]
- Berman, K.; Kwo, P.Y. Boceprevir, an NS3 protease inhibitor of HCV. Clin. Liver Dis. 2009, 13, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Kieffer, T.L.; Bartels, D.; Hanzelka, B.; Muh, U.; Welker, M.; Wincheringer, D.; Zhou, Y.; Chu, H.M.; Lin, C.; et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 2007, 132, 1767–1777. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Sarrazin, C.; Miller, J.S.; Welker, M.W.; Forestier, N.; Reesink, H.W.; Kwong, A.D.; Zeuzem, S. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology 2007, 46, 631–639. [Google Scholar] [CrossRef]
- Susser, S.; Welsch, C.; Wang, Y.; Zettler, M.; Domingues, F.S.; Karey, U.; Hughes, E.; Ralston, R.; Tong, X.; Herrmann, E.; et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 2009, 50, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Reesink, H.W.; Fanning, G.C.; Farha, K.A.; Weegink, C.; Van Vliet, A.; Van ’t Klooster, G.; Lenz, O.; Aharchi, F.; Marien, K.; Van Remoortere, P.; et al. Rapid HCV-RNA decline with once daily TMC435: A phase I study in healthy volunteers and hepatitis C patients. Gastroenterology 2010, 138, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.J.; Carroll, S.S.; Dimuzio, J.; Fandozzi, C.; Graham, D.J.; Hazuda, D.; Holloway, M.K.; Ludmerer, S.W.; McCauley, J.A.; McIntyre, C.J.; et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 2010, 54, 305–311. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Misialek, S.; Stevens, S.K.; Myszka, D.G.; Brandhuber, B.J.; Ballard, J.A.; Andrews, S.W.; Seiwert, S.D.; Kossen, K. Inhibition and binding kinetics of the hepatitis C virus NS3 protease inhibitor ITMN-191 reveals tight binding and slow dissociative behavior. Biochemistry 2009, 48, 2559–2568. [Google Scholar] [CrossRef]
- Bartels, D.J.; Zhou, Y.; Zhang, E.Z.; Marcial, M.; Byrn, R.A.; Pfeiffer, T.; Tigges, A.M.; Adiwijaya, B.S.; Lin, C.; Kwong, A.D.; et al. Natural prevalence of hepatitis C virus variants with decreased sensitivity to NS3.4A protease inhibitors in treatment-naive subjects. J. Infect. Dis. 2008, 198, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lennon, N.; Berlin, A.M.; Young, S.K.; Lee, B.; Heckerman, D.; Carlson, J.; Reyor, L.L.; et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology 2008, 48, 1769–1778. [Google Scholar] [CrossRef]
- Thibeault, D.; Bousquet, C.; Gingras, R.; Lagace, L.; Maurice, R.; White, P.W.; Lamarre, D. Sensitivity of NS3 serine proteases from hepatitis C virus genotypes 2 and 3 to the inhibitor BILN 2061. J. Virol. 2004, 78, 7352–7359. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Labrador, F.X.; Moya, A.; Gonzalez-Candelas, F. Mapping natural polymorphisms of hepatitis C virus NS3/4A protease and antiviral resistance to inhibitors in worldwide isolates. Antivir. Ther. 2008, 13, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Beyer, B.M.; Zhang, R.; Hong, Z.; Madison, V.; Malcolm, B.A. Effect of naturally occurring active site mutations on hepatitis C virus NS3 protease specificity. Proteins 2001, 43, 82–88. [Google Scholar] [CrossRef]
- Di Marco, S.; Rizzi, M.; Volpari, C.; Walsh, M.A.; Narjes, F.; Colarusso, S.; De Francesco, R.; Matassa, V.G.; Sollazzo, M. Inhibition of the hepatitis C virus NS3/4A protease. The crystal structures of two protease-inhibitor complexes. J. Biol. Chem. 2000, 275, 7152–7157. [Google Scholar] [CrossRef]
- Courcambeck, J.; Bouzidi, M.; Perbost, R.; Jouirou, B.; Amrani, N.; Cacoub, P.; Pepe, G.; Sabatier, J.M.; Halfon, P. Resistance of hepatitis C virus to NS3-4A protease inhibitors: Mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antivir. Ther. 2006, 11, 847–855. [Google Scholar] [CrossRef]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; Myers, J., Jr.; Weber, P.C.; Beyer, B.M.; Ingram, R.; et al. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3- [2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl] - 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 2007, 50, 2310–2318. [Google Scholar]
- Frecer, V.; Kabelac, M.; De Nardi, P.; Pricl, S.; Miertus, S. Structure-based design of inhibitors of NS3 serine protease of hepatitis C virus. J. Mol. Graph. Model. 2004, 22, 209–220. [Google Scholar] [CrossRef]
- McCown, M.F.; Rajyaguru, S.; Le Pogam, S.; Ali, S.; Jiang, W.R.; Kang, H.; Symons, J.; Cammack, N.; Najera, I. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob. Agents Chemother. 2008, 52, 1604–1612. [Google Scholar] [CrossRef]
- Ali, S.; Leveque, V.; Le Pogam, S.; Ma, H.; Philipp, F.; Inocencio, N.; Smith, M.; Alker, A.; Kang, H.; Najera, I.; et al. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 2008, 52, 4356–4369. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; King, M.S.; Kempf, D.J.; Lu, L.; Lim, H.B.; Krishnan, P.; Kati, W.; Middleton, T.; Molla, A. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob. Agents Chemother. 2008, 52, 1101–1110. [Google Scholar] [CrossRef]
- Carroll, S.S.; Ludmerer, S.; Handt, L.; Koeplinger, K.; Zhang, N.R.; Graham, D.; Davies, M.E.; MacCoss, M.; Hazuda, D.; Olsen, D.B. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob. Agents Chemother. 2009, 53, 926–934. [Google Scholar] [CrossRef]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef]
- Le Pogam, S.; Jiang, W.R.; Leveque, V.; Rajyaguru, S.; Ma, H.; Kang, H.; Jiang, S.; Singer, M.; Ali, S.; Klumpp, K.; et al. In vitro selected Con1 subgenomic replicons resistant to 2'-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 2006, 351, 349–359. [Google Scholar] [CrossRef]
- Pockros, P.J.; Nelson, D.; Godofsky, E.; Rodriguez-Torres, M.; Everson, G.T.; Fried, M.W.; Ghalib, R.; Harrison, S.; Nyberg, L.; Shiffman, M.L.; et al. R1626 plus peginterferon Alfa-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology 2008, 48, 385–397. [Google Scholar] [CrossRef]
- Gane, E.J.; Roberts, S.K.; Stedman, C.A.; Angus, P.W.; Ritchie, B.; Elston, R.; Ipe, D.; Morcos, P.N.; Baher, L.; Najera, I.; et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): A randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010, 376, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ng, K.K.; Cherney, M.M.; Chan, L.; Yannopoulos, C.G.; Bedard, J.; Morin, N.; Nguyen-Ba, N.; Alaoui-Ismaili, M.H.; Bethell, R.C.; et al. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003, 278, 9489–9495. [Google Scholar] [CrossRef] [PubMed]
- Kukolj, G.; McGibbon, G.A.; McKercher, G.; Marquis, M.; Lefebvre, S.; Thauvette, L.; Gauthier, J.; Goulet, S.; Poupart, M.A.; Beaulieu, P.L. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 2005, 280, 39260–39267. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.Q.; Yang, Y.; Harris, S.F.; Leveque, V.; Whittington, H.J.; Rajyaguru, S.; Ao-Ieong, G.; McCown, M.F.; Wong, A.; Giannetti, A.M.; et al. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. J. Biol. Chem. 2009, 284, 15517–15529. [Google Scholar] [CrossRef] [PubMed]
- Le Pogam, S.; Kang, H.; Harris, S.F.; Leveque, V.; Giannetti, A.M.; Ali, S.; Jiang, W.R.; Rajyaguru, S.; Tavares, G.; Oshiro, C.; et al. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 2006, 80, 6146–6154. [Google Scholar] [CrossRef]
- Pauwels, F.; Mostmans, W.; Quirynen, L.M.; van der Helm, L.; Boutton, C.W.; Rueff, A.S.; Cleiren, E.; Raboisson, P.; Surleraux, D.; Nyanguile, O.; et al. Binding-site identification and genotypic profiling of hepatitis C virus polymerase inhibitors. J. Virol. 2007, 81, 6909–6919. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Cheng, H.; Johann, S.; Mullen, S.; Chunduru, S.K.; Young, D.C.; Bard, J.; Chopra, R.; Krishnamurthy, G.; Mansour, T.; et al. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 2008, 52, 3327–3338. [Google Scholar] [CrossRef]
- Le Pogam, S.; Seshaadri, A.; Kosaka, A.; Chiu, S.; Kang, H.; Hu, S.; Rajyaguru, S.; Symons, J.; Cammack, N.; Najera, I. Existence of hepatitis C virus NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J. Antimicrob. Chemother. 2008, 61, 1205–1216. [Google Scholar] [CrossRef]
- Gaudieri, S.; Rauch, A.; Pfafferott, K.; Barnes, E.; Cheng, W.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; Mollison, L.; Baker, R.; et al. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2009, 49, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J.C.; Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008, 82, 1073–1083. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; O’Boyle, D., 2nd; Liu, M.; Nower, P.T.; Colonno, R.; Deshpande, M.S.; Snyder, L.B.; Martin, S.W.; St Laurent, D.R.; Serrano-Wu, M.H.; et al. Identification of hepatitis C virus NS5A inhibitors. J. Virol. 2010, 84, 482–491. [Google Scholar] [CrossRef]
- Fridell, R.A.; Qiu, D.; Wang, C.; Valera, L.; Gao, M. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 2010, 54, 3641–3650. [Google Scholar] [CrossRef]
- Yang, F.; Robotham, J.M.; Grise, H.; Frausto, S.; Madan, V.; Zayas, M.; Bartenschlager, R.; Robinson, M.; Greenstein, A.E.; Nag, A.; et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 2010, 6, e1001118. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Metral, V.; et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 2009, 49, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010, 53, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, V.; Di Stefano, R.; Ferraro, D.; Almasio, P.L.; Bonura, C.; Giglio, M.; Parisi, P.; Cappello, M.; Alaimo, G.; Craxi, A. HBV-DNA suppression and disease course in HBV cirrhosis patients on long-term lamivudine therapy. Antivir. Ther. 2005, 10, 431–439. [Google Scholar] [CrossRef]
- Erhardt, A.; Deterding, K.; Benhamou, Y.; Reiser, M.; Forns, X.; Pol, S.; Calleja, J.L.; Ross, S.; Spangenberg, H.C.; Garcia-Samaniego, J.; et al. Safety, pharmacokinetics and antiviral effect of BILB 1941, a novel hepatitis C virus RNA polymerase inhibitor, after 5 days oral treatment. Antivir. Ther. 2009, 14, 23–32. [Google Scholar] [CrossRef]
- Hirashima, S.; Suzuki, T.; Ishida, T.; Noji, S.; Yata, S.; Ando, I.; Komatsu, M.; Ikeda, S.; Hashimoto, H. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: Structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 2006, 49, 4721–4736. [Google Scholar] [CrossRef]
- Cooper, C.; Lawitz, E.J.; Ghali, P.; Rodriguez-Torres, M.; Anderson, F.H.; Lee, S.S.; Bedard, J.; Chauret, N.; Thibert, R.; Boivin, I.; et al. Evaluation of VCH-759 monotherapy in hepatitis C infection. J. Hepatol. 2009, 51, 39–46. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Bisbocci, M.; Bailey, C.; Bosserman, M.; Cellucci, A.; Forte, E.; Incitti, I.; Orsatti, L.; et al. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 2004, 78, 938–946. [Google Scholar] [CrossRef]
- McCown, M.F.; Rajyaguru, S.; Kular, S.; Cammack, N.; Najera, I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob. Agents Chemother. 2009, 53, 2129–2132. [Google Scholar] [CrossRef]

| Virus | Genomic Classification | Intracellular Reservoir | Mutation Rate* | Plasma Levels† | Recombination | Antiviral Drug Classes§ |
|---|---|---|---|---|---|---|
| HIV | Retrovirus | Proviral DNA | 10−5 | 103 − 106 | Major contribution to virus evolution in individuals and populations | Nucleoside RT inhibitors Nonnucleoside RT inhibitors Protease inhibitors Integrase inhibitors Fusion inhibitors CCR5 inhibitors |
| HBV | DS DNA virus with obligate RNA intermediate | Nuclear covalently-closed circular DNA (cccDNA) | 10−5 | 105 − 109 | Possible contribution to virus evolution within individuals | Interferon Nucleoside RT inhibitors |
| HCV | Positive single-stranded RNA virus | None | 10−4 − 10−5 | 104 − 107 | Possible contribution to virus evolution within individuals | Interferon + Ribavirin Protease inhibitors Nucleoside inhibitors Nonnucleoside inhibitors NS5A inhibitors Cyclophilin inhibitors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margeridon-Thermet, S.; Shafer, R.W. Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C. Viruses 2010, 2, 2696-2739. https://doi.org/10.3390/v2122696
Margeridon-Thermet S, Shafer RW. Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C. Viruses. 2010; 2(12):2696-2739. https://doi.org/10.3390/v2122696
Chicago/Turabian StyleMargeridon-Thermet, Severine, and Robert W. Shafer. 2010. "Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C" Viruses 2, no. 12: 2696-2739. https://doi.org/10.3390/v2122696
APA StyleMargeridon-Thermet, S., & Shafer, R. W. (2010). Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C. Viruses, 2(12), 2696-2739. https://doi.org/10.3390/v2122696