HIV-1 Entry, Inhibitors, and Resistance

Abstract
:1. Overview of HIV-1 Antiretroviral Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand Name | Generic Name | Class | Manufacturer | Approval Date |
|---|---|---|---|---|
| Emtriva | emtricitabine, FTC | NRTI | Gilead Sciences | 07/02/03 |
| Epivir | Lamivudine, 3TC | NRTI | GlaxoSmithKline | 11/17/95 |
| Hivid | Zalcitabine, ddC | NRTI | Hoffmann-LaRoche | 06/19/92 |
| Retrovir | Zidovudine, AZT | NRTI | GlaxoSmithKline | 03/19/87 |
| Videx | Didanosine, ddI | NRTI | Bristol-Myers Squibb | 10/09/91 |
| Viread | Tenofovir, TDF | NRTI | Gilead Sciences | 10/26/01 |
| Zerit | Stavudine, d4T | NRTI | Bristol-Myers Squibb | 06/24/94 |
| Ziagen | Abacavir, ABC | NRTI | GlaxoSmithKline | 12/17/98 |
| Intelence | Etravirine, ETV | NNRTI | Tibotec Therapeutics | 01/18/08 |
| Rescriptor | Delavirdine, DLV | NNRTI | Pfizer | 04/04/97 |
| Sustiva | Efavirenz, EVF | NNRTI | Bristol-Myers Squibb | 09/17/98 |
| Viramune | Nevirapine, NVP | NNRTI | Boehringer Ingelheim | 06/21/96 |
| Agenerase | Amprenavir, APV | PI | GlaxoSmithKline | 04/15/99 |
| Aptivus | Tipranavir, TPV | PI | Boehringer Ingelheim | 06/22/05 |
| Crixivan | Indinavir, IDV | PI | Merck | 03/13/96 |
| Invirase | Saquinavir, SQV | PI | Hoffmann-LaRoche | 12/06/95 |
| Lexiva | Fosamprenavir, APV | PI | GlaxoSmithKline | 10/20/03 |
| Norvir | Ritonavir, RTV | PI | Abbott Laboratories | 03/01/96 |
| Prezista | Darunavir | PI | Tibotec, Inc. | 06/23/06 |
| Reyataz | Atazanavir, ATV | PI | Bristol-Myers Squibb | 06/20/03 |
| Viracept | Nelfinavir, NFV | PI | Agouron | 03/14/97 |
| Fuzeon | Enfuvirtide, T-20 | Fusion | Hoffman-LaRoche and Trimeris | 03/13/03 |
| Selzentry | Maraviroc, MVC | CCR5/entry | Pfizer | 08/06/07 |
| Isentress | Raltegravir, RTV | Integrase | Merck | 10/12/07 |
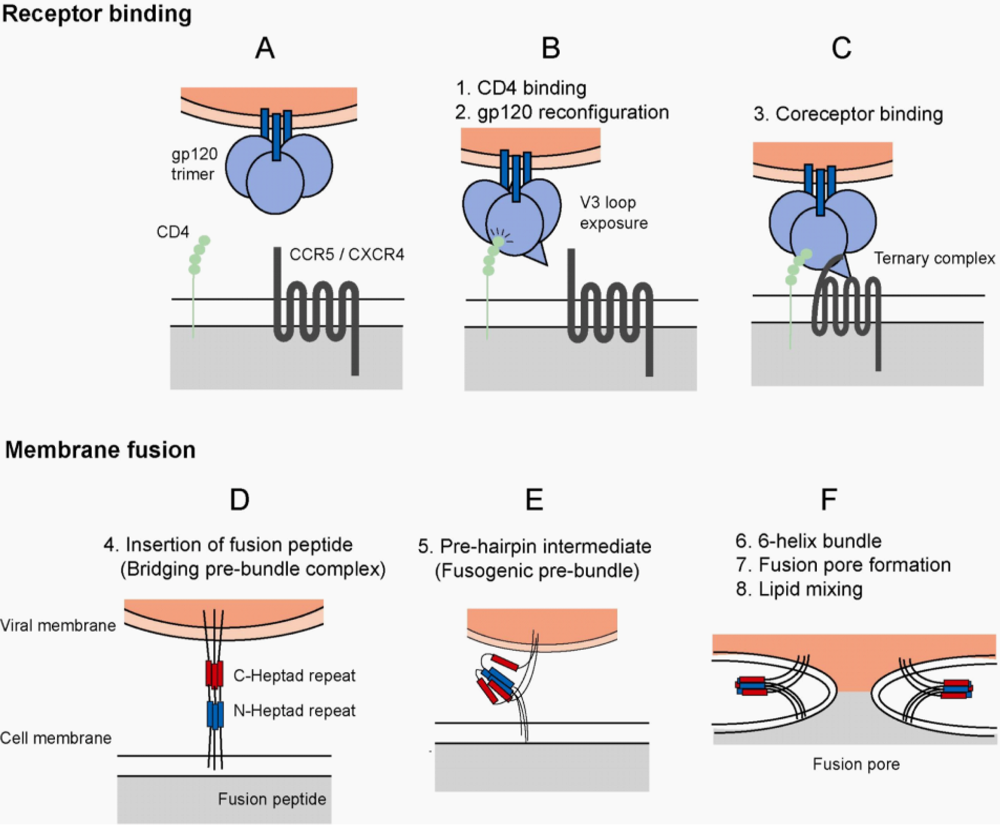
2. HIV-1 Envelope and Host Cell Entry

3. HIV-1 Receptors and Implications of Tropism
4. Interaction of HIV-1 Envelope and CCR5

5. Inhibition of HIV-1 Entry
5.1. Attachment Inhibitors
5.2. Fusion Inhibitors and Mechanisms of Resistance
| Compound | Mechanism | Status | Manufacturer |
|---|---|---|---|
| T-20 | Fusion inhibitor | Approved | Trimeris |
| T-1249 | Fusion inhibitor | Discontinued | Trimeris |
| C-34 | Fusion inhibitor | Preclinical only | --- |
| 5-Helix | Fusion inhibitor | --- | --- |
| Maraviroc | CCR5 antagonist | Approved | Pfizer |
| Vicriviroc (SCH-D) | CCR5 antagonist | Phase III | Schering-Plough |
| SCH-C | CCR5 antagonist | Phase I | Schering-Plough |
| AD101 | CCR5 antagonist | Preclinical only | Schering-Plough |
| Aplaviroc | CCR5 antagonist | Discontinued from Phase I | GlaxoSmithKline |
| TAK-652 | CCR5 antagonist | Phase I | Takeda |
| TAK-779 | CCR5 antagonist | Preclinical only | Takeda |
| INCB 9471 | CCR5 antagonist | Phase IIa | Incyte |
| AMD3100 | CXCR4 antagonist | Phase I/II | AnorMED |
| PRO-140 | Humanized anti-CCR5 monoconal antibody | Phase IIa | Progenics |
| PSC-RANTES | Chemokine analog | (Microbicide) | Gryphon |
| BMS-378806 | Attachment inhibitor | Phase IIa | Bristol-Myers Squibb |
| PRO-542 | CD4-Ig fusion | Discontinued | Progenics |
| TNX-355 | Anti-CD4 monoclonal antibody | Phase IIa | Biogen Idec; Tanox |
5.3. Chemokine Analogs
5.4. Small Molecule CCR5 Antagonists
6. Resistance to Coreceptor Inhibitors
6.1. Tropism Switching

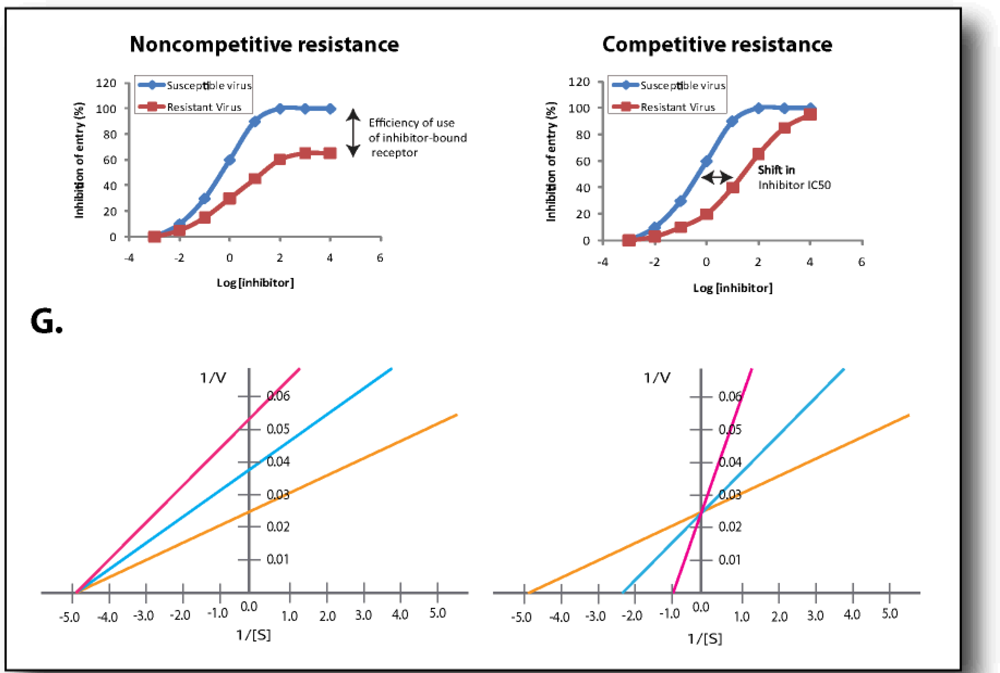
6.2. Competitive Resistance Model: Increased Coreceptor Affinity
6.3. Non-competitive Resistance Model: Utilization of Inhibitor-bound Coreceptor

| Resistant virus | V3 Loop resistance mutations (HXB2 numbering) | Reference |
|---|---|---|
| CC1/85 – Maraviroc | 316T | [136] |
| 323V | ||
| RU570 – Maraviroc | QAI deletion 315-17 | [136] |
| RU570 - Vicriviroc | 305R | [146] |
| 315Q | ||
| 319T | ||
| D101.12-Vicriviroc (CC1/85 derived) | 308P | [134] |
| CC101.19 – AD101 (CC1/85 derived) | 305R | [140] |
| 308P | ||
| 316V | ||
| 321E | ||
| Week 28 in vivo patient isolate- Vicriviroc | 305R | [141] |
| 306P | ||
| 307I | ||
| 316I | ||
| 318R | ||
| 319E |
7. Intrinsic Resistance to Entry Inhibitors
8. Implications of HIV-1 Entry Efficiency
9. Conclusions
Acknowledgments
References
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Bartlett, J.G.; Gallant, J.E.; Pham, P. Medical Management of HIV Infection, 15th ed; Knowledge Source Solutions: Baltimore, MD, USA, 2009. [Google Scholar]
- U.S. Food and Drug Administration. Approved HIV-1 Antiretrovirals . http://www.fda.gov (accessed December 3, 2009). [PubMed]
- Dalgleish, A.G.; Beverley, P.C.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.S.; Gowda, S.D.; Lifson, J.D.; Penhallow, R.C.; Bensch, K.G.; Engleman, E.G. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 1987, 49, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.; Sodroski, J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Doms, R.W.; Lamb, R.A.; Rose, J.K.; Helenius, A. Folding and assembly of viral membrane proteins. Virology 1993, 193, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Moss, B.; Doms, R.W. Folding, interaction with GRP78-BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J. Virol. 1991, 65, 2047–2055. [Google Scholar] [PubMed]
- Moulard, M.; Decroly, E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim. Biophys. Acta 2000, 1469, 121–132. [Google Scholar] [PubMed]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef]
- McDougal, J.S.; Nicholson, J.K.; Cross, G.D.; Cort, S.P.; Kennedy, M.S.; Mawle, A.C. Binding of the human retrovirus HTLV-III/LAV/ARV/HIV to the CD4 (T4) molecule: conformation dependence, epitope mapping, antibody inhibition, and potential for idiotypic mimicry. J. Immunol. 1986, 137, 2937–2944. [Google Scholar] [PubMed]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van, R.D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; Burton, D.R.; Dimitrov, D.S.; Sodroski, J.; Wyatt, R.; Nabel, G.J.; Kwong, P.D. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007, 445, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Fridell, R.A.; Aramori, I.; Ferguson, S.S.; Caron, M.G.; Cullen, B.R. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 co-receptor. EMBO J. 1997, 16, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, C.; Sodroski, J. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retroviruses 2000, 16, 741–749. [Google Scholar] [PubMed]
- Sattentau, Q.J.; Moore, J.P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Trkola, A.; Dragic, T.; Arthos, J.; Binley, J.M.; Olson, W.C.; Allaway, G.P.; Cheng-Mayer, C.; Robinson, J.; Maddon, P.J.; Moore, J.P. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 1996, 384, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, W.R. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell 1987, 50, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Harter, C.; James, P.; Bachi, T.; Semenza, G.; Brunner, J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the "fusion peptide". J. Biol. Chem. 1989, 264, 6459–6464. [Google Scholar] [PubMed]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000, 151, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Myszka, D.G.; Sweet, R.W.; Hensley, P.; Brigham-Burke, M.; Kwong, P.D.; Hendrickson, W.A.; Wyatt, R.; Sodroski, J.; Doyle, M.L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 9026–9031. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Wharton, S.A.; Calder, L.J.; Earl, P.L.; Moss, B.; Aliprandis, E.; Skehel, J.J.; Wiley, D.C. The ectodomain of HIV-1 env subunit gp41 forms a soluble, alpha-helical, rod-like oligomer in the absence of gp120 and the N-terminal fusion peptide. EMBO J. 1996, 15, 1507–1514. [Google Scholar] [PubMed]
- Chambers, P.; Pringle, C.R.; Easton, A.J. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990, 71, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L.; Mosialos, G.; Gilmore, T. Retroviral envelope glycoproteins contain a "leucine zipper"-like repeat. AIDS Res. Hum. Retroviruses 1990, 6, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, W.R.; Ball, J.M.; Garry, R.F.; Griffin, M.C.; Montelaro, R.C. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res. Hum. Retroviruses 1989, 5, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Byrn, R.A.; Marsters, S.A.; Gregory, T.; Groopman, J.E.; Capon, D.J. Blocking of HIV-1 infectivity by a soluble, secreted form of the CD4 antigen. Science 1987, 238, 1704–1707. [Google Scholar] [PubMed]
- Deen, K.C.; McDougal, J.S.; Inacker, R.; Folena-Wasserman, G.; Arthos, J.; Rosenberg, J.; Maddon, P.J.; Axel, R.; Sweet, R. W. A soluble form of CD4 (T4) protein inhibits AIDS virus infection. Nature 1988, 331, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Traunecker, A.; Luke, W.; Karjalainen, K. Soluble CD4 molecules neutralize human immunodeficiency virus type 1. Nature 1988, 331, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Hussey, R.E.; Richardson, N.E.; Kowalski, M.; Brown, N.R.; Chang, H.C.; Siliciano, R.F.; Dorfman, T.; Walker, B.; Sodroski, J.; Reinherz, E.L. A soluble CD4 protein selectively inhibits HIV replication and syncytium formation. Nature 1988, 331, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Bertonis, J.M.; Meier, W.; Johnson, V.A.; Costopoulos, D.S.; Liu, T.; Tizard, R.; Walker, B.D.; Hirsch, M.S.; Schooley, R.T. HIV infection is blocked in vitro by recombinant soluble CD4 . Nature 1988, 331, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Reimann, K.A.; DeLong, P.A.; Liu, T.; Fisher, R.A.; Letvin, N.L. Effect of recombinant soluble CD4 in rhesus monkeys infected with simian immunodeficiency virus of macaques. Nature 1989, 337, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Merigan, T.C.; Gaut, P.; Hirsch, M.S.; Holodniy, M.; Flynn, T.; Liu, S.; Byington, R.E.; Henochowicz, S.; Gubish, E. Recombinant soluble CD4 therapy in patients with the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. A phase I-II escalating dosage trial. Ann. Intern. Med. 1990, 112, 247–253. [Google Scholar] [PubMed]
- Traunecker, A.; Schneider, J.; Kiefer, H.; Karjalainen, K. Highly efficient neutralization of HIV with recombinant CD4-immunoglobulin molecules. Nature 1989, 339, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Daar, E.S.; Li, X.L.; Moudgil, T.; Ho, D.D. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc. Natl. Acad. Sci. U. S. A 1990, 87, 6574–6578. [Google Scholar] [CrossRef] [PubMed]
- Ivey-Hoyle, M.; Culp, J.S.; Chaikin, M.A.; Hellmig, B.D.; Matthews, T.J.; Sweet, R.W.; Rosenberg, M. Envelope glycoproteins from biologically diverse isolates of immunodeficiency viruses have widely different affinities for CD4. Proc. Natl. Acad. Sci. U. S. A 1991, 88, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.L.; Kahn, J.O.; Kaplan, L.D.; Groopman, J.E.; Volberding, P.A.; Amman, A.J.; Arri, C.J.; Bouvier, L.M.; Mordenti, J.; Izu, A.E. Phase 1 study of recombinant human CD4-immunoglobulin G therapy of patients with AIDS and AIDS-related complex . Antimicrob. Agents Chemother. 1991, 35, 2580–2586. [Google Scholar] [PubMed]
- Collier, A.C.; Coombs, R.W.; Katzenstein, D.; Holodniy, M.; Gibson, J.; Mordenti, J.; Izu, A.E.; Duliege, A.M.; Ammann, A.J.; Merigan, T. Safety, pharmacokinetics, and antiviral response of CD4-immunoglobulin G by intravenous bolus in AIDS and AIDS-related complex . J. Acquir. Immune. Defic. Syndr. Hum. Retrovirol. 1995, 10, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, J.E.; Gaudernack, G.; Vartdal, F. CD8+ T cells inhibit HIV replication in naturally infected CD4+ T cells. Evidence for a soluble inhibitor. J. Immunol. 1990, 144, 2961–2966. [Google Scholar] [PubMed]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar] [PubMed]
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hebert, C.A.; Horuk, R.; Matsushima, K.; Miller, L. H.; Oppenheim, J.J.; Power, C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000, 52, 145–176. [Google Scholar]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [PubMed]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.M.; Clark-Lewis, I.; Legler, D.F.; Loetscher, M.; Baggiolini, M.; Moser, B. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996, 382, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Farzan, M.; Choe, H.; Parolin, C.; Clark-Lewis, I.; Sodroski, J.; Springer, T.A. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996, 382, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [PubMed]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; Gerard, N.; Gerard, C.; Sodroski, J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di, M.P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; Davis, C.B.; Peiper, S.C.; Schall, T.J.; Littman, D.R.; Landau, N.R. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; Paxton, W.A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.M.; Deng, H.; Unutmaz, D.; Kewalramani, V.N.; Bastiani, L.; Gorny, M.K.; Zolla-Pazner, S.; Littman, D.R. Envelope glycoproteins from human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus can use human CCR5 as a coreceptor for viral entry and make direct CD4-dependent interactions with this chemokine receptor. J. Virol. 1997, 71, 6296–6304. [Google Scholar] [PubMed]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Hoffman, T.L.; Sharron, M.; Lee, B.; Yi, Y.; Choe, W.; Kolson, D.L.; Mitrovic, B.; Zhou, Y.; Faulds, D.; Collman, R.G.; Hesselgesser, J.; Horuk, R.; Doms, R.W. An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 1998, 72, 7934–7940. [Google Scholar] [PubMed]
- Farzan, M.; Choe, H.; Martin, K.; Marcon, L.; Hofmann, W.; Karlsson, G.; Sun, Y.; Barrett, P.; Marchand, N.; Sullivan, N.; Gerard, N.; Gerard, C.; Sodroski, J. Two orphan seven-transmembrane segment receptors which are expressed in CD4-positive cells support simian immunodeficiency virus infection. J. Exp. Med. 1997, 186, 405–411. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; Mackay, C.R.; Sodroski, J.; Gabuzda, D. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Alkhatib, G.; Peden, K.W.; Sharma, G.; Berger, E.A.; Farber, J.M. STRL33, A novel chemokine receptor-like protein, functions as a fusion cofactor for both macrophage-tropic and T cell line-tropic HIV-1. J. Exp. Med. 1997, 185, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Doms, R.W.; Fenyo, E.M.; Korber, B.T.; Littman, D.R.; Moore, J.P.; Sattentau, Q.J.; Schuitemaker, H.; Sodroski, J.; Weiss, R.A. A new classification for HIV-1. Nature 1998, 391, 240. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Tersmette, M.; de Goede, R.E.; Al, B.J.; Winkel, I.N.; Gruters, R.A.; Cuypers, H.T.; Huisman, H.G.; Miedema, F. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J. Virol. 1988, 62, 2026–2032. [Google Scholar] [PubMed]
- Schuitemaker, H.; Koot, M.; Kootstra, N.A.; Dercksen, M.W.; de Goede, R.E.; van Steenwijk, R.P.; Lange, J.M.; Schattenkerk, J.K.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar] [PubMed]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; Donfield, S.; Vlahov, D.; Kaslow, R.; Saah, A.; Rinaldo, C.; Detels, R.; O'Brien, S.J. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; Kunstman, K.; Erickson, D.; Dragon, E.; Landau, N.R.; Phair, J.; Ho, D.D.; Koup, R.A. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 1996, 2, 1240–1243. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.L.; Chang, G.; Louie, L.G.; Mascola, J.R.; Dondero, D.; Birx, D.L.; Sheppard, H.W. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nat. Med. 1997, 3, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; Muyldermans, G.; Verhofstede, C.; Burtonboy, G.; Georges, M.; Imai, T.; Rana, S.; Yi, Y.; Smyth, R.J.; Collman, R.G.; Doms, R.W.; Vassart, G.; Parmentier, M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, P.A.; Buckler-White, A.; Alkhatib, G.; Spalding, T.; Kubofcik, J.; Combadiere, C.; Weissman, D.; Cohen, O.; Rubbert, A.; Lam, G.; Vaccarezza, M.; Kennedy, P.E.; Kumaraswami, V.; Giorgi, J.V.; Detels, R.; Hunter, J.; Chopek, M.; Berger, E.A.; Fauci, A.S.; Nutman, T.B.; Murphy, P.M. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol. Med. 1997, 3, 23–36. [Google Scholar] [PubMed]
- An, P.; Martin, M.P.; Nelson, G.W.; Carrington, M.; Smith, M.W.; Gong, K.; Vlahov, D.; O'Brien, S.J.; Winkler, C.A. Influence of CCR5 promoter haplotypes on AIDS progression in African-Americans. AIDS 2000, 14, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Dean, M.; Smith, M.W.; Winkler, C.; Gerrard, B.; Michael, N.L.; Lee, B.; Doms, R.W.; Margolick, J.; Buchbinder, S.; Goedert, J.J.; O'Brien, T.R.; Hilgartner, M.W.; Vlahov, D.; O'Brien, S.J.; Carrington, M. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science 1998, 282, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Zimmerman, P.A.; Guignard, F.; Kleeberger, C.A.; Leitman, S.F.; Murphy, P.M. CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS). Lancet 1998, 352, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Ometto, L.; Bertorelle, R.; Mainardi, M.; Zanchetta, M.; Tognazzo, S.; Rampon, O.; Ruga, E.; Chieco-Bianchi, L.; De, R.A. Polymorphisms in the CCR5 promoter region influence disease progression in perinatally human immunodeficiency virus type 1-infected children. J. Infect. Dis. 2001, 183, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Astemborski, J.; Bashirova, A.; Mosbruger, T.; Greer, S.; Witt, M.D.; Goedert, J.J.; Hilgartner, M.; Majeske, A.; O'Brien, S.J.; Thomas, D.L.; Carrington, M. Genetic protection against hepatitis B virus conferred by CCR5Delta32: Evidence that CCR5 contributes to viral persistence. J. Virol. 2007, 81, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hartley, O.; Klasse, P.J.; Sattentau, Q.J.; Moore, J.P. V3: HIV's switch-hitter. AIDS Res. Hum. Retroviruses 2005, 21, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Nabatov, A.A.; Pollakis, G.; Linnemann, T.; Kliphius, A.; Chalaby, M.I.; Paxton, W.A. Intrapatient alterations in the human immunodeficiency virus type 1 gp120 V1V2 and V3 regions differentially modulate coreceptor usage, virus inhibition by CC/CXC chemokines, soluble CD4, and the b12 and 2G12 monoclonal antibodies. J. Virol. 2004, 78, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Doranz, B.J.; Lu, Z.H.; Rucker, J.; Zhang, T.Y.; Sharron, M.; Cen, Y.H.; Wang, Z.X.; Guo, H.H.; Du, J.G.; Accavitti, M.A.; Doms, R.W.; Peiper, S.C. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J. Virol. 1997, 71, 6305–6314. [Google Scholar] [PubMed]
- Picard, L.; Simmons, G.; Power, C.A.; Meyer, A.; Weiss, R.A.; Clapham, P.R. Multiple extracellular domains of CCR-5 contribute to human immunodeficiency virus type 1 entry and fusion. J. Virol. 1997, 71, 5003–5011. [Google Scholar] [PubMed]
- Cormier, E.G.; Dragic, T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 2002, 76, 8953–8957. [Google Scholar] [CrossRef] [PubMed]
- Farzan, M.; Mirzabekov, T.; Kolchinsky, P.; Wyatt, R.; Cayabyab, M.; Gerard, N.P.; Gerard, C.; Sodroski, J.; Choe, H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 1999, 96, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Tang, M.; Zhang, M.Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; Wyatt, R.; Kwong, P.D. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Sharron, M.; Blanpain, C.; Doranz, B.J.; Vakili, J.; Setoh, P.; Berg, E.; Liu, G.; Guy, H.R.; Durell, S.R.; Parmentier, M.; Chang, C.N.; Price, K.; Tsang, M.; Doms, R.W. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 1999, 274, 9617–9626. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Vanderwinden, J.M.; Cihak, J.; Wittamer, V.; Le, P.E.; Issafras, H.; Stangassinger, M.; Vassart, G.; Marullo, S.; Schlndorff, D.; Parmentier, M.; Mack, M. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol. Biol. Cell 2002, 13, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Falcon, P.; Rodriguez-Frade, J.M.; Serrano, A.; Juan, D.; del, S.A.; Soriano, S.F.; Roncal, F.; Gomez, L.; Valencia, A.; Martinez, A.; Mellado, M. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 2004, 5, 216–223. [Google Scholar] [CrossRef]
- Issafras, H.; Angers, S.; Bulenger, S.; Blanpain, C.; Parmentier, M.; Labbe-Jullie, C.; Bouvier, M.; Marullo, S. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J. Biol. Chem. 2002, 277, 34666–34673. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.; Rose, R.; Yamanaka, G.; Robinson, B.; Li, C.B.; Fridell, R.; Deminie, C.; Demers, G.; Yang, Z.; Zadjura, L.; Meanwell, N.; Colonno, R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 11013–11018. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.T.; Fan, L.; Nowicka-Sans, B.; McAuliffe, B.; Li, C.B.; Yamanaka, G.; Zhou, N.; Fang, H.; Dicker, I.; Dalterio, R.; Gong, Y.F.; Wang, T.; Yin, Z.; Ueda, Y.; Matiskella, J.; Kadow, J.; Clapham, P.; Robinson, J.; Colonno, R.; Lin, P.F. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J. Virol. 2006, 80, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Malashkevich, V.N.; Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 9134–9139. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.B.; Dutch, R.E.; Lamb, R.A. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology 1998, 248, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.; Oas, T.; McDanal, C.; Bolognesi, D.; Matthews, T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 10537–10541. [Google Scholar] [CrossRef] [PubMed]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; Matthews, T.; Johnson, M.R.; Nowak, M.A.; Shaw, G.M.; Saag, M.S. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Henry, K.; O'Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron Jr., J.J.; Chung, J.; Demasi, R.; Donatacci, L.; Drobnes, C.; Delehanty, J.; Salgo, M. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America . N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sista, P.; Giguel, F.; Greenberg, M.; Kuritzkes, D.R. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 2004, 78, 4628–4637. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Lee, F.H.; Miamidian, J.L.; Jabara, C.B.; Juntilla, M.M.; Doms, R.W. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 2005, 79, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar] [CrossRef] [PubMed]
- Labrosse, B.; Morand-Joubert, L.; Goubard, A.; Rochas, S.; Labernardiere, J.L.; Pacanowski, J.; Meynard, J.L.; Hance, A.J.; Clavel, F.; Mammano, F. Role of the envelope genetic context in the development of enfuvirtide resistance in human immunodeficiency virus type 1-infected patients. J. Virol. 2006, 80, 8807–8819. [Google Scholar] [CrossRef] [PubMed]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O'Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Gallo, S.A.; Ahmad, N.; Miamidian, J.L.; Harvey, P.E.; Sharron, M.; Pohlmann, S.; Sfakianos, J.N.; Derdeyn, C.A.; Blumenthal, R.; Hunter, E.; Doms, R.W. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16249–16254. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Miamidian, J.L.; Biscone, M.J.; Lee, F.H.; Ahmad, N.; Pierson, T.C.; Doms, R.W. Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and entry inhibitor sensitivity. J. Virol. 2004, 78, 5476–5485. [Google Scholar] [CrossRef] [PubMed]
- Biscone, M.J.; Miamidian, J.L.; Muchiri, J.M.; Baik, S.S.; Lee, F.H.; Doms, R.W.; Reeves, J.D. Functional impact of HIV coreceptor-binding site mutations. Virology 2006, 351, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, C.L.; Kollmann, C.; Giguel, F.; Chou, T.C.; Hirsch, M.S. Strong in vitro synergy between the fusion inhibitor T-20 and the CXCR4 blocker AMD-3100. J. Acquir. Immune. Defic. Syndr. 2000, 25, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Sorensen, M.; Fung, M.; Schooley, R.T. Synergistic in vitro antiretroviral activity of a humanized monoclonal anti-CD4 antibody (TNX-355) and enfuvirtide (T-20). Antimicrob. Agents Chemother. 2006, 50, 2231–2233. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.A.; Thompson, D.A.; Rosenfield, S.I.; Maddon, P.J.; Dragic, T.; Olson, W.C. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J. Infect. Dis. 2001, 183, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Clapham, P.R.; Picard, L.; Offord, R.E.; Rosenkilde, M.M.; Schwartz, T.W.; Buser, R.; Wells, T.N.; Proudfoot, A.E. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 1997, 276, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Townson, J.R.; Graham, G.J.; Landau, N.R.; Rasala, B.; Nibbs, R.J. Aminooxypentane addition to the chemokine macrophage inflammatory protein-1alpha P increases receptor affinities and HIV inhibition. J. Biol. Chem. 2000, 275, 39254–39261. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Doranz, B.J.; Bondue, A.; Govaerts, C.; De, L.A.; Vassart, G.; Doms, R.W.; Proudfoot, A.; Parmentier, M. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J. Biol. Chem. 2003, 278, 5179–5187. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Martin, A.; Nieto, M.; Sanchez-Madrid, F.; Proudfoot, A.E.; Wells, T.N.; Martinez, A.; Mellado, M. Similarities and differences in RA. J. Cell Biol. 1999, 144, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Signoret, N.; Pelchen-Matthews, A.; Mack, M.; Proudfoot, A.E.; Marsh, M. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell Biol. 2000, 151, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Gall, S.L.; Schwartz, O.; Salamero, J.; Montes, M.; Loetscher, P.; Baggiolini, M.; Virelizier, J.L.; Arenzana-Seisdedos, F. HIV coreceptor downregulation as antiviral principle: SDF-1alpha-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J. Exp. Med. 1997, 186, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Pastore, C.; Picchio, G.R.; Galimi, F.; Fish, R.; Hartley, O.; Offord, R.E.; Mosier, D.E. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 2003, 47, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.M.; Mariani, R.; Holland, A.U.; Hope, T.J.; Landau, N.R. Association of chemokine-mediated block to HIV entry with coreceptor internalization. J. Biol. Chem. 2002, 277, 17291–17299. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Luckow, B.; Nelson, P.J.; Cihak, J.; Simmons, G.; Clapham, P.R.; Signoret, N.; Marsh, M.; Stangassinger, M.; Borlat, F.; Wells, T.N.; Schlondorff, D.; Proudfoot, A.E. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 1998, 187, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Hartley, O.; Gaertner, H.; Wilken, J.; Thompson, D.; Fish, R.; Ramos, A.; Pastore, C.; Dufour, B.; Cerini, F.; Melotti, A.; Heveker, N.; Picard, L.; Alizon, M.; Mosier, D.; Kent, S.; Offord, R. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 16460–16465. [Google Scholar] [CrossRef] [PubMed]
- Lederman, M.M.; Veazey, R.S.; Offord, R.; Mosier, D.E.; Dufour, J.; Mefford, M.; Piatak Jr., M.; Lifson, J.D.; Salkowitz, J.R.; Rodriguez, B.; Blauvelt, A.; Hartley, O. Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5 . Science 2004, 306, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Lobritz, M.A.; Ratcliff, A.N.; Marozsan, A.J.; Dudley, D.M.; Arts, E.J. XVIII International HIV Drug Resistance Workshop (Abstract #12). 9-13 6; Fort Myers, FL, USA, 2009. [Google Scholar]
- Vila-Coro, A.J.; Mellado, M.; Martin de, A.A.; Martinez, A.; Rodriguez-Frade, J.M. Characterization of RA. J. Immunol. 1999, 163, 3037–3044. [Google Scholar] [PubMed]
- Marozsan, A.J.; Torre, V.S.; Johnson, M.; Ball, S.C.; Cross, J.V.; Templeton, D.J.; Quinones-Mateu, M.E.; Offord, R.E.; Arts, E.J. Mechanisms involved in stimulation of human immunodeficiency virus type 1 replication by aminooxypentane RANTES. J. Virol. 2001, 75, 8624–8638. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Muesing, M.A.; Proudfoot, A.E.; Power, C.A.; Moore, J.P.; Trkola, A. Enhancement of human immunodeficiency virus type 1 infection by the CC-chemokine RANTES is independent of the mechanism of virus-cell fusion. J. Virol. 1999, 73, 684–694. [Google Scholar] [PubMed]
- Roscic-Mrkic, B.; Fischer, M.; Leemann, C.; Manrique, A.; Gordon, C.J.; Moore, J.P.; Proudfoot, A.E.; Trkola, A. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood 2003, 102, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Trkola, A.; Thompson, D.A.; Cormier, E.G.; Kajumo, F.A.; Maxwell, E.; Lin, S.W.; Ying, W.; Smith, S.O.; Sakmar, T.P.; Moore, J.P. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5639–5644. [Google Scholar] [CrossRef] [PubMed]
- Tsamis, F.; Gavrilov, S.; Kajumo, F.; Seibert, C.; Kuhmann, S.; Ketas, T.; Trkola, A.; Palani, A.; Clader, J.W.; Tagat, J.R.; McCombie, S.; Baroudy, B.; Moore, J.P.; Sakmar, T.P.; Dragic, T. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 2003, 77, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S.; Klasse, P.J.; Ketas, T.J.; Reeves, J.D.; Piatak Jr., M.; Kunstman, K.; Kuhmann, S.E.; Marx, P.A.; Lifson, J.D.; Dufour, J.; Mefford, M.; Pandrea, I.; Wolinsky, S.M.; Doms, R.W.; DeMartino, J.A.; Siciliano, S.J.; Lyons, K.; Springer, M.S.; Moore, J.P. Use of a small molecule CCR5 inhibitor in macaques to treat simian immunodeficiency virus infection or prevent simian-human immunodeficiency virus infection. J. Exp. Med. 2003, 198, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S.; Springer, M.S.; Marx, P.A.; Dufour, J.; Klasse, P.J.; Moore, J.P. Protection of macaques from vaginal SHIV challenge by an orally delivered CCR5 inhibitor. Nat. Med. 2005, 11, 1293–1294. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; Webster, R.; Armour, D.; Price, D.; Stammen, B.; Wood, A.; Perros, M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Kondru, R.; Zhang, J.; Ji, C.; Mirzadegan, T.; Rotstein, D.; Sankuratri, S.; Dioszegi, M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 2008, 73, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; Montana, J.B.; McHale, M.; Sullivan, J.; Ridgway, C.; Felstead, S.; Dunne, M.W.; van der Ryst, E.; Mayer, H. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; Bellos, N.; Saag, M.; Cooper, D.A.; Westby, M.; Tawadrous, M.; Sullivan, J.F.; Ridgway, C.; Dunne, M.W.; Felstead, S.; Mayer, H.; van der Ryst, E. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Suleiman, J.; Diaz, R.; Madruga, J.; DeJesus, E.; Zingman, B.; Slim, J.; Case, N.; Dunkle, L. 49th ICAAC (Abstract H-923) . 2009. [Google Scholar]
- Crabb, C. GlaxoSmithKline ends aplaviroc trials. AIDS 2006, 20, 641. [Google Scholar] [CrossRef] [PubMed]
- Marozsan, A.J.; Kuhmann, S.E.; Morgan, T.; Herrera, C.; Rivera-Troche, E.; Xu, S.; Baroudy, B. M.; Strizki, J.; Moore, J.P. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology 2005, 338, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Trkola, A.; Kuhmann, S.E.; Strizki, J.M.; Maxwell, E.; Ketas, T.; Morgan, T.; Pugach, P.; Xu, S.; Wojcik, L.; Tagat, J.; Palani, A.; Shapiro, S.; Clader, J.W.; McCombie, S.; Reyes, G.R.; Baroudy, B.M.; Moore, J.P. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 2007, 81, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Miyake, H.; Wang, X.; Okamoto, M.; Takashima, K. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob. Agents Chemother. 2007, 51, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.; Goodrich, J.; Fatkenheuer, G.; Clotet, B.; Clumeck, N.; Sullivan, J.; Westby, M.; van der Ryst, E.; Mayer, H. A double-blind, placebo-controlled trial of maraviroc in treatment-experienced patients infected with non-R5 HIV-1. J. Infect. Dis. 2009, 199, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Kuhmann, S.E.; Pugach, P.; Kunstman, K.J.; Taylor, J.; Stanfield, R.L.; Snyder, A.; Strizki, J.M.; Riley, J.; Baroudy, B.M.; Wilson, I.A.; Korber, B.T.; Wolinsky, S.M.; Moore, J.P. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J. Virol. 2004, 78, 2790–2807. [Google Scholar] [CrossRef] [PubMed]
- Tsibris, A.M.; Sagar, M.; Gulick, R.M.; Su, Z.; Hughes, M.; Greaves, W.; Subramanian, M.; Flexner, C.; Giguel, F.; Leopold, K.E.; Coakley, E.; Kuritzkes, D.R. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J. Virol. 2008, 82, 8210–8214. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.M.; Wentzel, J.L.; Lalonde, M.S.; Veazey, R.S.; Arts, E.J. Selection of a simian-human immunodeficiency virus strain resistant to a vaginal microbicide in macaques. J. Virol. 2009, 83, 5067–5076. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Mosley, M.; Lewis, M.; Simpson, P.; Toma, J.; Huang, J. XVI International HIV Drug Resistance Workshop (Abstract 10). 6 12-16; Barbados, 2007. [Google Scholar]
- Pugach, P.; Marozsan, A.J.; Ketas, T.J.; Landes, E.L.; Moore, J.P.; Kuhmann, S.E. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology 2007, 361, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.G.; Ketas, T.J.; Klasse, P.J.; Moore, J.P. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 5318–5323. [Google Scholar] [CrossRef] [PubMed]
- Ogert, R.A.; Wojcik, L.; Buontempo, C.; Ba, L.; Buontempo, P.; Ralston, R.; Strizki, J.; Howe, J.A. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology 2008, 373, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.G.; Marozsan, A.J.; Matet, A.; Snyder, A.D.; Arts, E.J.; Kuhmann, S.E.; Moore, J.P. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss . PLoS. Pathog. 2007, 3, e79. [Google Scholar] [CrossRef] [PubMed]
- Torre, V.S.; Marozsan, A.J.; Albright, J.L.; Collins, K.R.; Hartley, O.; Offord, R.E.; Quinones-Mateu, M.E.; Arts, E.J. Variable sensitivity of CCR5-tropic human immunodeficiency virus type 1 isolates to inhibition by RANTES analogs. J. Virol. 2000, 74, 4868–4876. [Google Scholar] [CrossRef] [PubMed]
- Labrosse, B.; Labernardiere, J.L.; Dam, E.; Trouplin, V.; Skrabal, K.; Clavel, F.; Mammano, F. Baseline susceptibility of primary human immunodeficiency virus type 1 to entry inhibitors. J. Virol. 2003, 77, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Safarian, D.; Carnec, X.; Tsamis, F.; Kajumo, F.; Dragic, T. An anti-CCR5 monoclonal antibody and small molecule CCR5 antagonists synergize by inhibiting different stages of human immunodeficiency virus type 1 entry. Virology 2006, 352, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Strizki, J.M.; Xu, S.; Wagner, N.E.; Wojcik, L.; Liu, J.; Hou, Y.; Endres, M.; Palani, A.; Shapiro, S.; Clader, J.W.; Greenlee, W.J.; Tagat, J.R.; McCombie, S.; Cox, K.; Fawzi, A.B.; Chou, C.C.; Pugliese-Sivo, C.; Davies, L.; Moreno, M.E.; Ho, D.D.; Trkola, A.; Stoddart, C.A.; Moore, J.P.; Reyes, G.R.; Baroudy, B.M. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Platt, E.J.; Durnin, J.P.; Kabat, D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 2005, 79, 4347–4356. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, S.R.; Markosyan, R.M.; Eadon, M.T.; Moore, J.P.; Melikyan, G.B.; Cohen, F.S. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured as an intermediate of membrane fusion. J. Virol. 2005, 79, 11161–11169. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Picchio, G.R.; Gulizia, R.J.; Sabbe, R.; Poignard, P.; Picard, L.; Offord, R.E.; Thompson, D.A.; Wilken, J. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J. Virol. 1999, 73, 3544–3550. [Google Scholar] [PubMed]
- Lobritz, M.A.; Marozsan, A.J.; Troyer, R.M.; Arts, E.J. Natural variation in the V3 crown of human immunodeficiency virus type 1 affects replicative fitness and entry inhibitor sensitivity. J. Virol. 2007, 81, 8258–8269. [Google Scholar] [CrossRef] [PubMed]
- Trkola, A.; Paxton, W.A.; Monard, S.P.; Hoxie, J.A.; Siani, M.A.; Thompson, D.A.; Wu, L.; Mackay, C.R.; Horuk, R.; Moore, J.P. Genetic subtype-independent inhibition of human immunodeficiency virus type 1 replication by CC and CXC chemokines. J. Virol. 1998, 72, 396–404. [Google Scholar] [PubMed]
- Koning, F.A.; Kwa, D.; Boeser-Nunnink, B.; Dekker, J.; Vingerhoed, J.; Hiemstra, H.; Schuitemaker, H. Decreasing sensitivity to RANTES (regulated on activation, normally T cell-expressed and -secreted) neutralization of CC chemokine receptor 5-using, non-syncytium-inducing virus variants in the course of human immunodeficiency virus type 1 infection. J. Infect. Dis. 2003, 188, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Kuhmann, S.E.; Platt, E.J.; Kozak, S.L.; Kabat, D. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J. Virol. 2000, 74, 7005–7015. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kurteva, S.; Ren, X.; Lee, S.; Sodroski, J. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 2005, 79, 12132–12147. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kurteva, S.; Ren, X.; Lee, S.; Sodroski, J. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J. Virol. 2006, 80, 4388–4395. [Google Scholar] [CrossRef] [PubMed]
- Layne, S.P.; Merges, M.J.; Dembo, M.; Spouge, J.L.; Nara, P.L. HIV requires multiple gp120 molecules for CD4-mediated infection. Nature 1990, 346, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.; Klasse, P.J.; Kibler, C.W.; Michael, E.; Moore, J.P.; Beddows, S. Dominant-negative effect of hetero-oligomerization on the function of the human immunodeficiency virus type 1 envelope glycoprotein complex. Virology 2006, 351, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Rangel, H.R.; Weber, J.; Chakraborty, B.; Gutierrez, A.; Marotta, M.L.; Mirza, M.; Kiser, P.; Martinez, M.A.; Este, J.A.; Quinones-Mateu, M.E. Role of the human immunodeficiency virus type 1 envelope gene in viral fitness. J. Virol. 2003, 77, 9069–9073. [Google Scholar] [CrossRef] [PubMed]
- Marozsan, A.J.; Moore, D.M.; Lobritz, M.A.; Fraundorf, E.; Abraha, A.; Reeves, J.D.; Arts, E.J. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J. Virol. 2005, 79, 7121–7134. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.S.; Hahn, B.H.; Gibbons, J.; Li, Y.; Parks, E.S.; Parks, W.P.; Shaw, G.M. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature 1988, 334, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Mateu, M.E.; Ball, S.C.; Marozsan, A.J.; Torre, V.S.; Albright, J.L.; Vanham, G.; van Der, G.G.; Colebunders, R.L.; Arts, E.J. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J. Virol. 2000, 74, 9222–9233. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; Lobritz, M.A.; Bailey, J.R.; Johnston, S.; Nguyen, S.; Lee, B.; Chou, T.; Siliciano, R.F.; Markowitz, M.; Arts, E.J. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS. Pathog. 2009, 5, e1000377. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Lobritz, M.A.; Ratcliff, A.N.; Arts, E.J. HIV-1 Entry, Inhibitors, and Resistance. Viruses 2010, 2, 1069-1105. https://doi.org/10.3390/v2051069
Lobritz MA, Ratcliff AN, Arts EJ. HIV-1 Entry, Inhibitors, and Resistance. Viruses. 2010; 2(5):1069-1105. https://doi.org/10.3390/v2051069
Chicago/Turabian StyleLobritz, Michael A., Annette N. Ratcliff, and Eric J. Arts. 2010. "HIV-1 Entry, Inhibitors, and Resistance" Viruses 2, no. 5: 1069-1105. https://doi.org/10.3390/v2051069
APA StyleLobritz, M. A., Ratcliff, A. N., & Arts, E. J. (2010). HIV-1 Entry, Inhibitors, and Resistance. Viruses, 2(5), 1069-1105. https://doi.org/10.3390/v2051069