Resistance to Integrase Inhibitors

Abstract
:
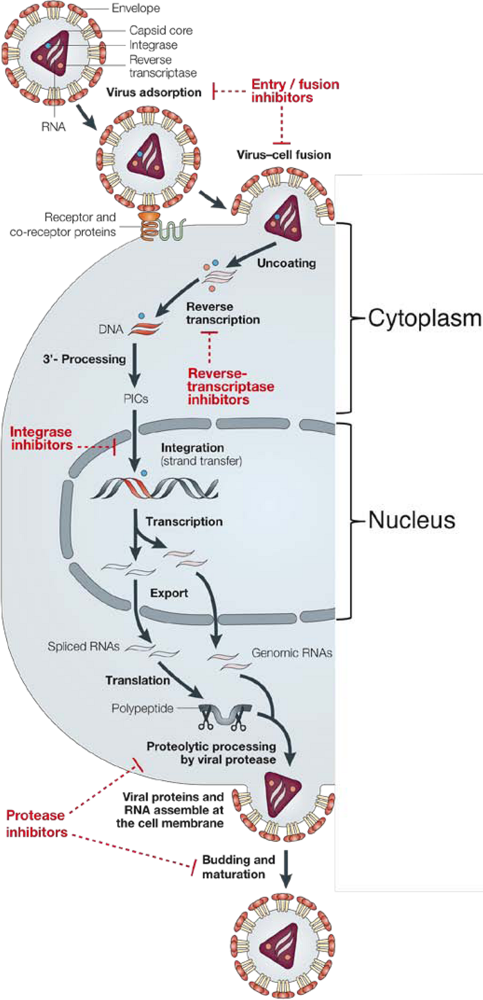
1. Background


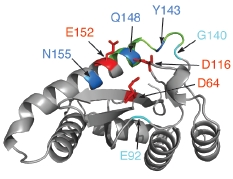
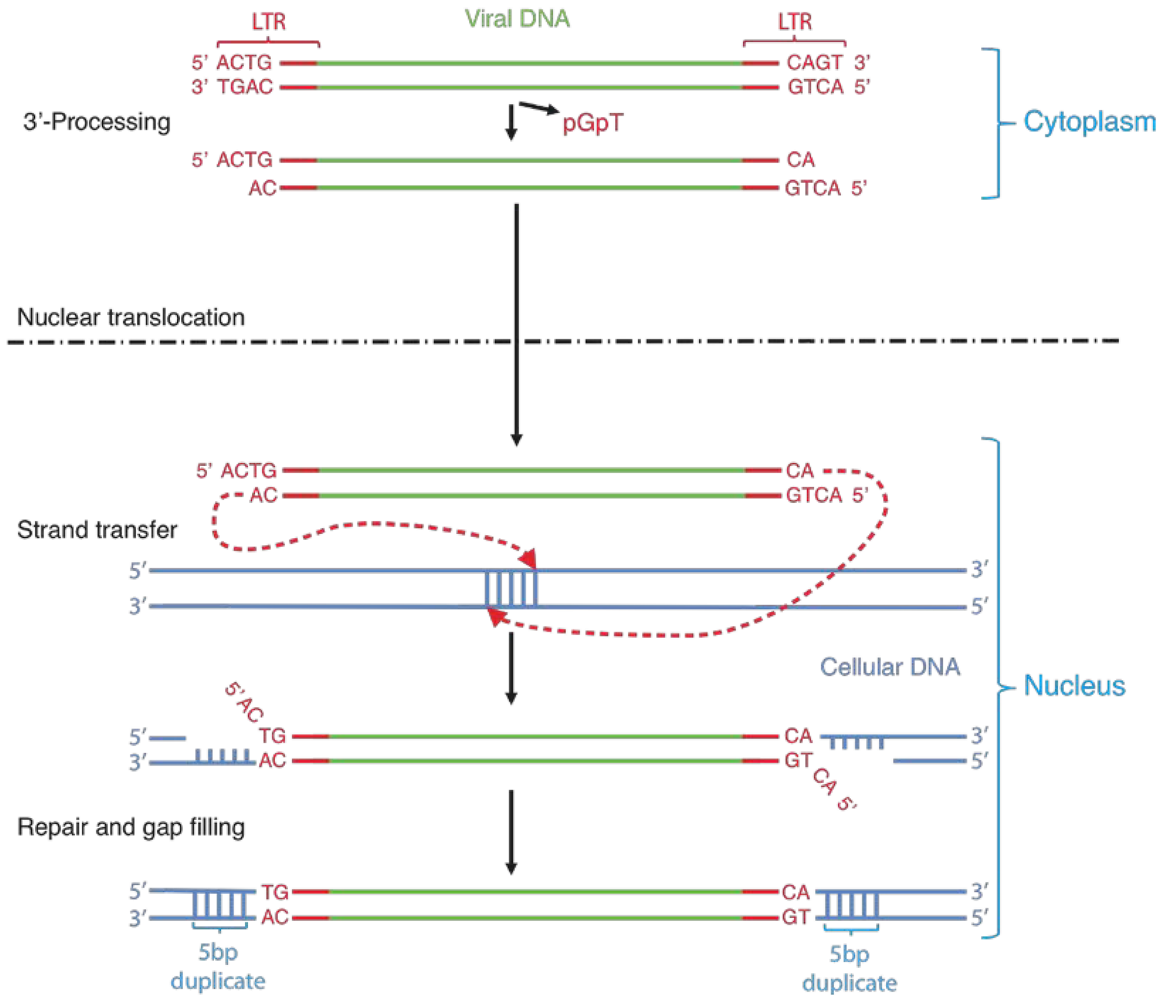
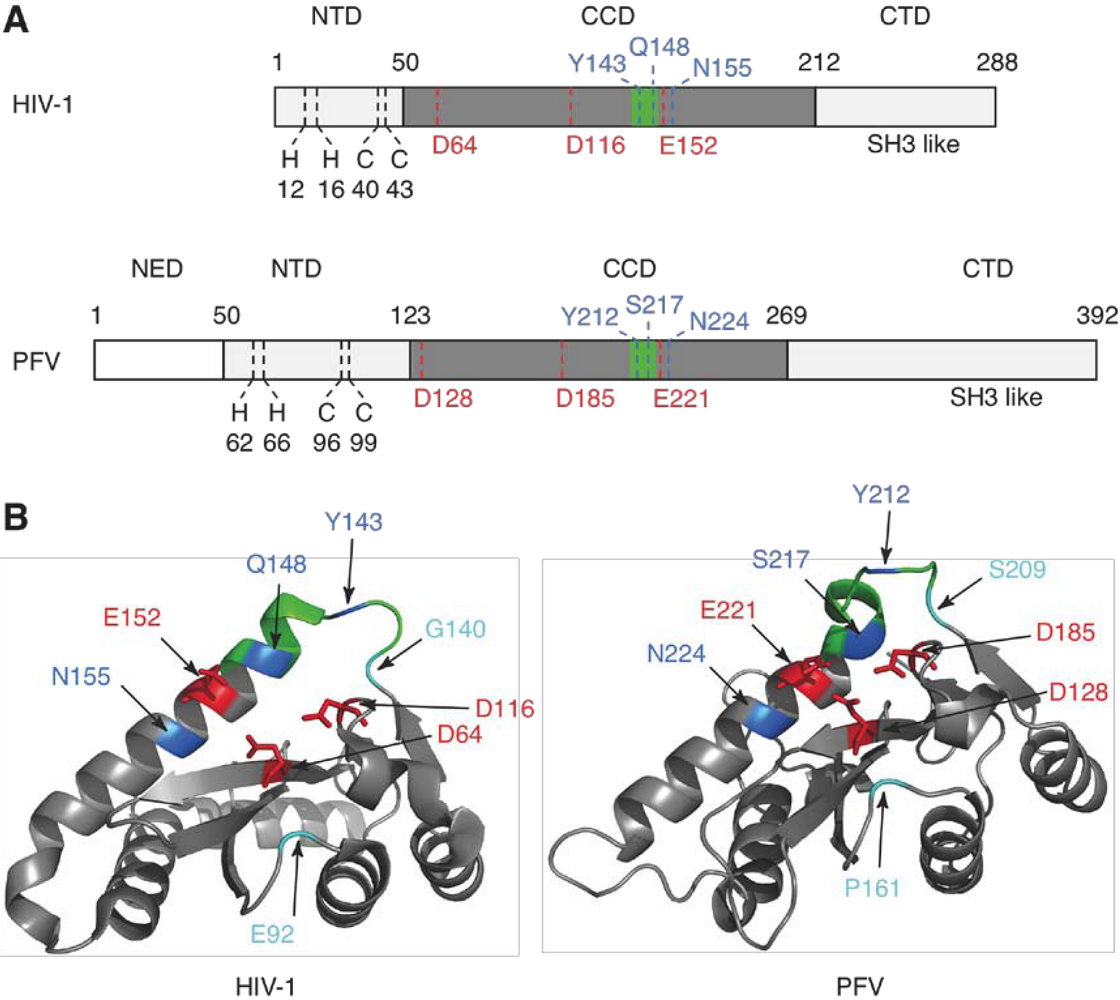
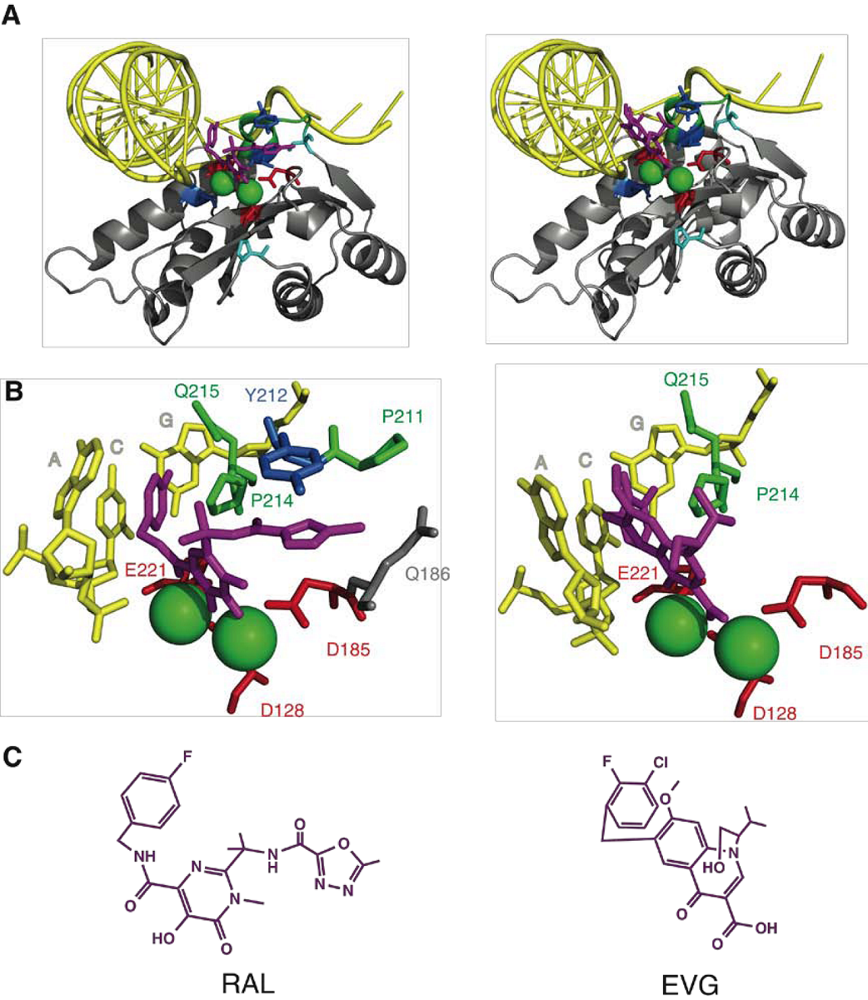
2. Integrase structure


3. Integrase inhibitors: historical overview
4. Raltegravir

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Company | Structure | Anti-INa | Anti-HIVb | T1/2 | Status | Ref. |
|---|---|---|---|---|---|---|---|
| Raltegravir MK-0518 Isenstress® | Merck & Co. |  | 2-7 | 8.9 | 1; 7-12 | FDA Approved October 2007 | [35,47,48,49] |
| Elvitegravir JTK-303 GS-9137 | Japan Tobacco Inc. and Gilead Sciences |  | 7 | 1.7 | 3; 9* | Phase III | [35,47] |
| S/GSK-1349572 | ViiV-Healthcare and Shionogi & Co. Ltd |  | 2.7 | 2 | 14 | Phase IIb | [35,50] |

5. IN mutations conferring RAL resistance
6. Elvitegravir
| IN | Virus | |||||||
|---|---|---|---|---|---|---|---|---|
| Mutations | 3’-P | ST | Rce | Selection | RC | Refs | ||
| H51Y | ++ | EVG | [76] | |||||
| H51Y/E92Q/S147G | + | EVG | [76] | |||||
| H51Y/E92Q/S147G/E157Q | ++ | EVG | [76] | |||||
| T66A | + | ++ | RAL/EVG | ++ | [63,77,78] | |||
| T66I | +++* | +++* | 51-102X | DKA/EVG | ++* | [29,38,79,80,81,82,83,84,85,86,87] | ||
| T66I/E92Q | - | EVG | [85,86] | |||||
| L74M | +++ | +++ | 1X1,2 | DKA/EVG | [29,80,84,85,88] | |||
| E92Q | +++* | +++* | 4-5X1,2 | DKA/RAL/EVG | ++ | [66,76,78,88] | ||
| E92Q/S147G | + | EVG | [76] | |||||
| E92Q/N155H | RAL | ++* | [72,89] | |||||
| E138K/Q148HRK | DKA/RAL | ++ | [72,87,90] | |||||
| G140S | +++* | + | 1X1,2 | DKA/RAL | ++ | [61,71,78,89,90,91,92,93] | ||
| G140A | ++ | + | 2-3X1 | RAL | [71] | |||
| G140S/Q148H | ++* | +++* | >50X1,2 | RAL | +++ | [61,66,72,94] | ||
| G140S/Q148K | ++ | + | 10-25X1 | DKA/RAL | ++ | [71,72,90] | ||
| G140S/Q148R | - | + | >25X1 | DKA/RAL | ++ | [56,72,87] | ||
| G140A/Q148H | - | + | >25X1 | RAL | + | [71,72] | ||
| G140A/Q148K | - | - | 2-3X1 | RAL | ++ | [71,72] | ||
| G140A/Q148R | + | + | >25X1 | RAL | ++ | [71,72,95] | ||
| G140S/Q148H/S230N | RAL | +++ | [89] | |||||
| Y143R | + | ++ | >50X1 | RAL | + | [56,62,93,96] | ||
| Y143C | + | + | >50X1 | RAL | + | [62] | ||
| Y143R/G163R | RAL | ++ | [89] | |||||
| S147G | + | EVG | [76] | |||||
| Q148K | - | +* | 50X1* | DKA/RAL/EVG | + | [28,29,71,72,87,90] | ||
| Q148R | +* | + | 2-3X1 | DKA/RAL/EVG | ++ | [71,72,78,85,86,87,90,95,97] | ||
| Q148H | - | +* | 2-3X1,2 | RAL | ++* | [61,71,72,78,89,93] | ||
| Q148H/N155H | RAL | + | [89] | |||||
| Q148K/G163R | RAL | [87] | ||||||
| N155H | +++* | +++* | 5-10X1,2 | DKA/RAL | ++ | [29,61,66,72,78,86,87,89,93,94,95,97,98] | ||
| E157Q | ++ | RAL/EVG | [76,99] | |||||
| S230R | +++ | +++ | DKA/EVG | [80,85] | ||||
7. Second generation INSTI
8. Conclusions
Acknowledgments
References
- Delelis, O.; Carayon, K.; Saib, A.; Deprez, E.; Mouscadet, J.F. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, H.; Hayashi, T.; Takahashi, T.; Miyano, M.; Kannagi, M.; Masuda, T. Augmentation of reverse transcription by integrase through an interaction with host factor, SIP1/Gemin2 Is critical for HIV-1 infection . PLoS One 2009, 4, e7825. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug. Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef]
- Jaskolski, M.; Alexandratos, J.N.; Bujacz, G.; Wlodawer, A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009, 276, 2926–2946. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.K.; Davies, D.R. Structure and function of HIV-1 integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef]
- Faure, A.; Calmels, C.; Desjobert, C.; Castroviejo, M.; Caumont-Sarcos, A.; Tarrago-Litvak, L.; Litvak, S.; Parissi, V. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005, 33, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Van Maele, B.; Debyser, Z. HIV-1 integration: an interplay between HIV-1 integrase, cellular and viral proteins. AIDS Rev. 2005, 7, 26–43. [Google Scholar] [PubMed]
- Van Maele, B.; Busschots, K.; Vandekerckhove, L.; Christ, F.; Debyser, Z. Cellular co-factors of HIV-1 integration. Trends Biochem. Sci. 2006, 31, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Darlix, J.L.; Cimarelli, A. Analysis of the viral elements required in the nuclear import of HIV-1 DNA. J. Virol. 2010, 84, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; Hughes, S.H. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication . PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R. Targeting HIV-1 DNA integration by swapping tethers. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2735–2736. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Mizuuchi, K.; Bushman, F.D.; Engelman, A. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 1991, 19, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Fesen, M.R.; Kohn, K.W.; Leteurtre, F.; Pommier, Y. Inhibitors of human immunodeficiency virus integrase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, G.; Im, G.J.; Brackmann, K.; Grandgenett, D. Concerted integration of retrovirus-like DNA by human immunodeficiency virus type 1 integrase. J. Virol. 1995, 69, 6090–6097. [Google Scholar] [PubMed]
- Nowotny, M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009, 10, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [PubMed]
- Greenwald, J.; Le, V.; Butler, S.L.; Bushman, F.D.; Choe, S. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 1999, 38, 8892–8898. [Google Scholar] [CrossRef] [PubMed]
- Maignan, S.; Guilloteau, J.P.; Zhou-Liu, Q.; Clement-Mella, C.; Mikol, V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J. Mol. Biol. 1998, 282, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Krucinski, J.; Miercke, L.J.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Ling, H.; Yang, W.; Craigie, R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001, 20, 7333–7343. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Gao, K.; Bushman, F.D.; Yeager, M. Single-particle image reconstruction of a tetramer of HIV integrase bound to DNA. J. Mol. Biol. 2007, 366, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wielens, J.; Crosby, I.T.; Chalmers, D.K. A three-dimensional model of the human immunodeficiency virus type 1 integration complex. J. Comput. Aided Mol. Des. 2005, 19, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Ambrosio, A.L.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17308–17313. [Google Scholar] [CrossRef] [PubMed]
- Fitzkee, N.C.; Masse, J.E.; Shen, Y.; Davies, D.R.; Bax, A. Solution conformation and dynamics of the HIV-1 integrase core domain. J. Biol. Chem. 2010, 285, 18072–18084. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Weber, I.T.; Harrison, R.W.; Leis, J. Identification of amino acids in HIV-1 and avian sarcoma virus integrase subsites required for specific recognition of the long terminal repeat Ends. J. Biol. Chem. 2006, 281, 4173–4182. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Santos, W.; Pais, G.C.; Marchand, C.; Amin, R.; Burke Jr., T.R.; Verdine, G.; Pommier, Y. Integration requires a specific interaction of the donor DNA terminal 5'-cytosine with glutamine 148 of the HIV-1 integrase flexible loop . J. Biol. Chem. 2006, 281, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Limon, A.; Ghory, H.Z.; Engelman, A. Genetic analyses of DNA-binding mutants in the catalytic core domain of human immunodeficiency virus type 1 integrase. J. Virol. 2005, 79, 2493–2505. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Marchand, C.; Mott, B.T.; Bain, A.; Thomas, C.J.; Pommier, Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 2008, 47, 9345–9354. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Craigie, R. Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J. 1998, 17, 5832–5843. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, M.L.; Arbildua, J.J.; Monasterio, O.; Toledo, H.; Leon, O. Role of the 207-218 peptide region of Moloney murine leukemia virus integrase in enzyme catalysis. Arch. Biochem. Biophys. 2010, 495, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Gupta, S.S.; Hare, S.; Helander, A.; Roversi, P.; McClure, M.; Cherepanov, P. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res. 2009, 37, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.; Maddali, K.; Métifiot, M.; Pommier, Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr. Top. Med. Chem. 2009, 9, 1016–1037. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, K.; Serrao, E.; Odde, S.; Neamati, N. HIV-1 integrase inhibitors: 2007-2008 update . Med. Res. Rev. 2010, In press. [Google Scholar]
- Liao, C.; Marchand, C.; Burke Jr., T.R.; Jr.; Pommier, Y.; Nicklaus, M.C. Authentic HIV-1 Integrase Inhibitors . Future Med. Chem. 2010, In press. [Google Scholar]
- Robinson Jr., W.E.; Reinecke, M.G.; Abdel-Malek, S.; Jia, Q.; Chow, S.A. Inhibitors of HIV-1 replication [corrected; erratum to be published] that inhibit HIV integrase. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6326–6331. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; Miller, M.D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Andreola, M.L. Therapeutic potential of peptide motifs against HIV-1 reverse transcriptase and integrase. Curr. Pharm. Des. 2009, 15, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Jing, N.; Xu, X. Rational drug design of DNA oligonucleotides as HIV inhibitors. Curr. Drug. Targets Infect. Disord. 2001, 1, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ojwang, J.O.; Buckheit, R.W.; Pommier, Y.; Mazumder, A.; De Vreese, K.; Este, J.A.; Reymen, D.; Pallansch, L.A.; Lackman-Smith, C.; Wallace, T.L.; et al. T30177, an oligonucleotide stabilized by an intramolecular guanosine octet, is a potent inhibitor of laboratory strains and clinical isolates of human immunodeficiency virus type 1 . Antimicrob. Agents Chemother. 1995, 39, 2426–2435. [Google Scholar] [PubMed]
- Jing, N.; Marchand, C.; Guan, Y.; Liu, J.; Pallansch, L.; Lackman-Smith, C.; De Clercq, E.; Pommier, Y. Structure-activity of inhibition of HIV-1 integrase and virus replication by G-quartet oligonucleotides. DNA Cell Biol. 2001, 20, 499–508. [Google Scholar] [PubMed]
- Cherepanov, P.; Este, J.A.; Rando, R.F.; Ojwang, J.O.; Reekmans, G.; Steinfeld, R.; David, G.; De Clercq, E.; Debyser, Z. Mode of interaction of G-quartets with the integrase of human immunodeficiency virus type 1. Mol. Pharmacol. 1997, 52, 771–780. [Google Scholar] [PubMed]
- Este, J.A.; Cabrera, C.; Schols, D.; Cherepanov, P.; Gutierrez, A.; Witvrouw, M.; Pannecouque, C.; Debyser, Z.; Rando, R.F.; Clotet, B.; Desmyter, J.; De Clercq, E. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol. Pharmacol. 1998, 53, 340–345. [Google Scholar] [PubMed]
- Semenova, E.A.; Marchand, C.; Pommier, Y. HIV-1 integrase inhibitors: update and perspectives. Adv. Pharmacol. 2008, 56, 199–228. [Google Scholar] [PubMed]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; Wai, J.S.; Young, S.; Vacca, J.; Hazuda, D.J. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 6661–6666. [Google Scholar] [CrossRef] [PubMed]
- Serrao, E.; Odde, S.; Ramkumar, K.; Neamati, N. Raltegravir, elvitegravir, and metoogravir: the birth of "me-too" HIV-1 integrase inhibitors. Retrovirology 2009, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Correll, T.; Klibanov, O.M. Integrase inhibitors: a new treatment option for patients with human immunodeficiency virus infection. Pharmacotherapy 2008, 28, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.R.; Gorzynski, J.E.; Drobysheva, D.; Bassit, L.; Schinazi, R.F. Raltegravir is a potent inhibitor of XMRV, a virus implicated in prostate cancer and chronic fatigue syndrome . PLoS One 2010, 5, e9948. [Google Scholar] [CrossRef] [PubMed]
- Vandeckerckhove, L. GSK-1349572, a novel integrase inhibitor for the treatment of HIV infection. Curr. Opin. Investig. Drugs 2010, 11, 203–212. [Google Scholar] [PubMed]
- Pommier, Y.; Cherfils, J. Interfacial protein inhibition: a nature's paradigm for drug discovery. Trends Pharmacol. Sci. 2005, 28, 136–145. [Google Scholar]
- FDA notifications. FDA approves raltegravir for HIV-1 treatment-naive patients . AIDS Alert 2009, 24, 106–107. [Google Scholar] [PubMed]
- Cocohoba, J. The SWITCHMRK studies: substitution of lopinavir/ritonavir with raltegravir in HIV-positive individuals. Expert Rev. Anti. Infect. Ther. 2009, 7, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Emery, S.; Winston, A. Raltegravir: a new choice in HIV and new chances for research. Lancet 2009, 374, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Soriano, V.; de Mendoza, C. New therapeutic strategies for raltegravir. J. Antimicrob. Chemother. 2010, 65, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Lennox, J.L.; DeJesus, E.; Lazzarin, A.; Pollard, R.B.; Madruga, J.V.; Berger, D.S.; Zhao, J.; Xu, X.; Williams-Diaz, A.; Rodgers, A.J.; Barnard, R.J.; Miller, M.D.; DiNubile, M.J.; Nguyen, B.Y.; Leavitt, R.; Sklar, P. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet 2009, 374, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Nguyen, B.Y.; Gotuzzo, E.; Mendo, F.; Ratanasuwan, W.; Kovacs, C.; Prada, G.; Morales-Ramirez, J.O.; Crumpacker, C.S.; Isaacs, R.D.; Gilde, L.R.; Wan, H.; Miller, M.D.; Wenning, L.A.; Teppler, H. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J. Acquir. Immune Defic. Syndr. 2007, 46, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Young, S.D.; Guare Jr., J.P.; Anthony, N.J.; Gomez, R.P.; Wai, J.S.; Vacca, J.P.; Handt, L.; Motzel, S.L.; Klein, H.J.; Dornadula, G.; Danovich, R.M.; Witmer, M.V.; Wilson, K.A.; Tussey, L.; Schleif, W.A.; Gabryelski, L.S.; Jin, L.; Miller,M.D.; Casimiro, D.R.; Emini, E.A.; Shiver, J.W. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques . Science 2004, 305, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Gupta, S.; Danovich, R.; Hazuda, D.; Miller, M.; Witmer, M.; Petropoulos, C.J.; Huang, W. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 2009, 83, 11440–11446. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Steigbigel, R.T.; Gatell, J.M.; Rockstroh, J.K.; Katlama, C.; Yeni, P.; Lazzarin, A.; Clotet, B.; Kumar, P.N.; Eron, J.E.; Schechter, M.; Markowitz, M.; Loutfy, M.R.; Lennox, J.L.; Zhao, J.; Chen, J.; Ryan, D.M.; Rhodes, R.R.; Killar, J.A.; Gilde, L.R.; Strohmaier, K.M.; Meibohm, A.R.; Miller, M.D.; Hazuda, D.J.; Nessly, M.L.; DiNubile, M.J.; Isaac,s R.D.; Teppler, H.; Nguyen, B.Y. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection . N. Engl. J. Med. 2008, 359, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Malet, I.; Na, L.; Tchertanov, L.; Calvez, V.; Marcelin, A.G.; Subra, F.; Deprez, E.; Mouscadet, J.F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009, 37, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Thierry, S.; Subra, F.; Simon, F.; Malet, I.; Alloui, C.; Sayon, S.; Calvez, V.; Deprez, E.; Marcelin, A.G.; Tchertanov, L.; Mouscadet, J.F. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob. Agents Chemother. 2010, 54, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, N.; Kubo, M.; Maeda, Y.; Ohashi, T.; Yamamoto, N.; Kannagi, M.; Masuda, T. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells. J. Virol. 2000, 74, 4795–4806. [Google Scholar] [CrossRef] [PubMed]
- Low, A.; Prada, N.; Topper, M.; Vaida, F.; Castor, D.; Mohri, H.; Hazuda, D.; Muesing, M.; Markowitz, M. Natural polymorphisms of human immunodeficiency virus type 1 integrase and inherent susceptibilities to a panel of integrase inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4275–4282. [Google Scholar] [CrossRef] [PubMed]
- Loizidou, E.Z.; Kousiappa, I.; Zeinalipour-Yazdi, C.D.; Van de Vijver, D.A.; Kostrikis, L.G. Implications of HIV-1 M group polymorphisms on integrase inhibitor efficacy and resistance: genetic and structural in silico analyses. Biochemistry 2009, 48, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Malet, I.; Delelis, O.; Valantin, M.A.; Montes, B.; Soulie, C.; Wirden, M.; Tchertanov, L.; Peytavin, G.; Reynes, J.; Mouscadet, J.F.; Katlama, C.; Calvez, V.; Marcelin, A.G. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Ceccherini-Silberstein, F.; Malet, I.; D'Arrigo, R.; Antinori, A.; Marcelin, A.G.; Perno, C.F. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009, 11, 17–29. [Google Scholar] [PubMed]
- Sichtig, N.; Sierra, S.; Kaiser, R.; Daumer, M.; Reuter, S.; Schulter, E.; Altmann, A.; Fatkenheuer, G.; Dittmer, U.; Pfister, H.; Esser, S. Evolution of raltegravir resistance during therapy. J. Antimicrob. Chemother. 2009, 64, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C.; Laureillard, D.; Piketty, C.; Tisserand, P.; Batisse, D.; Karmochkine, M.; Si-Mohamed, A.; Weiss, L. High frequency of integrase Q148R minority variants in HIV-infected patients naive of integrase inhibitors. AIDS 2010, 24, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Quercia, R.; Dam, E.; Perez-Bercoff, D.; Clavel, F. Selective-advantage profile of human immunodeficiency virus type 1 integrase mutants explains in vivo evolution of raltegravir resistance genotypes. J. Virol. 2009, 83, 10245–10249. [Google Scholar] [CrossRef] [PubMed]
- Métifiot, M.; Maddali, K.; Naumova, A.; Zhang, X.; Marchand, C.; Pommier, Y. Biochemical and Pharmacological Analyses of HIV-1 Integrase Flexible Loop Mutants Resistant to Raltegravir. Biochemistry 2010, 49, 3715–3722. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Karmochkine, M.; Huang, W.; Weiss, L.; Petropoulos, C.; Charpentier, C. Longitudinal analysis of raltegravir susceptibility and integrase replication capacity of HIV-1 during virologic failure. Antimicrob. Agents Chemother. 2009, 53, 4522–4524. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.; Van Wesenbeeck, L.; Breilh, D.; Reigadas, S.; Anies, G.; Van Baelen, K.; Morlat, P.; Neau, D.; Dupon, M.; Wittkop, L.; Fleury, H.; Masquelier, B. HIV-1 resistance patterns to integrase inhibitors in antiretroviral-experienced patients with virological failure on raltegravir-containing regimens. J. Antimicrob. Chemother. 2010, 65, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Zolopa, A.R.; Berger, D.S.; Lampiris, H.; Zhong, L.; Chuck, S.L.; Enejosa, J.V.; Kearney, B.P.; Cheng, A.K. Activity of Elvitegravir, a Once-Daily Integrase Inhibitor, against Resistant HIV Type 1: Results of a Phase 2, Randomized, Controlled, Dose-Ranging Clinical Trial. J. Infect. Dis. 2010, 201, 814–822. [Google Scholar] [CrossRef] [PubMed]
- DeJesus, E.; Berger, D.; Markowitz, M.; Cohen, C.; Hawkins, T.; Ruane, P. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J. Acquir. Immune Defic. Syndr. 2006, 43, 1–5. [Google Scholar] [CrossRef]
- Shimura, K.; Kodama, E.; Sakagami, Y.; Matsuzaki, Y.; Watanabe, W.; Yamataka, K.; Watanabe, Y.; Ohata, Y.; Doi, S.; Sato, M.; Kano, M.; Ikeda, S.; Matsuoka, M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Gerton, J.L.; Ohgi, S.; Olsen, M.; DeRisi, J.; Brown, P.O. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J. Virol. 1998, 72, 5046–5055. [Google Scholar] [PubMed]
- Charpentier, C.; Karmochkine, M.; Laureillard, D.; Tisserand, P.; Bélec, L.; Weiss, L.; Si-Mohamed, A.; Piketty, C. Drug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapy. HIV Med. 2008, 9, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, T.; Sato, A.; Fujishita, T.; Fujiwara, T. S-1360: in vitro activity of a new HIV-1 integrase inhibitor in clinical development . 2002; In Presented at the 9th Conference on Retrovirures and Opportunistic Infections. [Google Scholar]
- Fikkert, V.; Van Maele, B.; Vercammen, J.; Hantson, A.; Van Remoortel, B.; Michiels, M.; Gurnari, C.; Pannecouque, C.; De Maeyer, M.; Engelborghs, Y.; De Clercq, E.; Debyser, Z.; Witvrouw, M. Development of resistance against diketo derivatives of human immunodeficiency virus type 1 by progressive accumulation of integrase mutations. J. Virol. 2003, 77, 11459–11470. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Barr, R.; Zhang, X.; Pais, G.C.; Marchand, C.; Pommier, Y.; Burke Jr., T.R. Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two-long-terminal-repeat-circle junctions. J. Virol. 2004, 78, 3210–3222. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Robinson Jr., W.E. Human immunodeficiency virus type 1 (HIV-1) integrase: resistance to diketo acid integrase inhibitors impairs HIV-1 replication and integration and confers cross-resistance to L-chicoric acid . J. Virol. 2004, 78, 5835–5847. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Anthony, N.J.; Gomez, R.P.; Jolly, S.M.; Wai, J.S.; Zhuang, L.; Fisher, T.E.; Embrey, M.; Guare Jr., J.P.; Egbertson, M.S.; Vacca, J.P.; Huff, J.R.; Felock, P.J.; Witmer, M.V.; Stillmock, K.A.; Danovich, R.; Grobler, J.; Miller, M.D.; Espeseth, A.S.; Jin, L.; Chen, I.W.; Lin, J.H.; Kassahun, K.; Ellis, J.D.; Wong, B.K.; Xu, W.; Pearson, P.G.; Schleif, W.A.; Cortese, R.; Emini, E.; Summa, V.; Holloway, M.K.; Young, S.D. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11233–11238. [Google Scholar] [CrossRef] [PubMed]
- Fikkert, V.; Hombrouck, A.; Van Remoortel, B.; De Maeyer, M.; Pannecouque, C.; De Clercq, E.; Debyser, Z.; Witvrouw, M. Multiple mutations in human immunodeficiency virus-1 integrase confer resistance to the clinical trial drug S-1360. Aids 2004, 18, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Goethals, O.; Clayton, R.; Van Ginderen, M.; Vereycken, I.; Wagemans, E.; Geluykens, P.; Dockx, K.; Strijbos, R.; Smits, V.; Vos, A.; Meersseman, G.; Jochmans, D.; Vermeire, K.; Schols, D.; Hallenberger, S.; Hertogs, K. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008, 82, 10366–10374. [Google Scholar] [CrossRef] [PubMed]
- Dicker, I.B.; Terry, B.; Lin, Z.; Li, Z.; Bollini, S.; Samanta, H.K.; Gali, V.; Walker, M.A.; Krystal, M.R. Biochemical Analysis of HIV-1 Integrase Variants Resistant to Strand Transfer Inhibitors. J. Biol. Chem. 2008, 283, 23599–23609. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakahara, K.; Seki, T.; Miki, S.; Kawauchi, S.; Suyama, A.; Wakasa-Morimoto, C.; Kodama, M.; Endoh, T.; Oosugi, E.; Matsushita, Y.; Murai, H.; Fujishita, T.; Yoshinaga, T.; Garvey, E.; Foster, S.; Underwood, M.; Johns, B.; Sato, A.; Fujiwara, T. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral Res. 2008, 80, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hombrouck, A.; Voet, A.; Van Remoortel, B.; Desadeleer,, C.; De Maeyer, M.; Debyser, Z.; Witvrouw, M. Mutations in HIV-1 Integrase Confer Resistance to the Naphthyridine L-870,810 and Cross Resistance to the Clinical Trial drug GS-9137 . Antimicrob. Agents Chemother. 2008, 52, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Buzón, M.; Dalmau, J.; Puertas, M.; Puig, J.; Clotet, B.; Martinez-Picado, J. The HIV-1 integrase genotype strongly predicts raltegravir susceptibility but not viral fitness of primary virus isolates. Aids 2009, 24, 17–25. [Google Scholar] [CrossRef]
- Nakahara, K.; Wakasa-Morimoto, C.; Kobayashi, M.; Miki, S.; Noshi, T.; Seki, T.; Kanamori-Koyama, M.; Kawauchi, S.; Suyama, A.; Fujishita, T.; Yoshinaga, T.; Garvey, E.; Johns, B.; Foster, S.; Underwood, M.; Sato, A.; Fujiwara, T. Secondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antiviral Res. 2008, 81, 141–146. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Lee, D.J.; Reinke, R.A.; Victoria, J.G.; Beale, K.; Robinson Jr., W.E. Human immunodeficiency virus type-1 integrase containing a glycine to serine mutation at position 140 is attenuated for catalysis and resistant to integrase inhibitors . Virology 2003, 306, 147–161. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Robinson Jr., W.E. Resistance to the anti-human immunodeficiency virus type 1 compound L-chicoric acid results from a single mutation at amino acid 140 of integrase . J. Virol. 1998, 72, 8420–8424. [Google Scholar] [PubMed]
- Baldanti, F.; Paolucci, S.; Gulminetti, R.; Brandolini, M.; Barbarini, G.; Maserati, R. Early emergence of raltegravir resistance mutations in patients receiving HAART salvage regimens. J. Med. Virol. 2009, 82, 116–122. [Google Scholar] [CrossRef]
- Malet, I.; Delelis, O.; Soulie, C.; Wirden, M.; Tchertanov, L.; Mottaz, P.; Peytavin, G.; Katlama, C.; Mouscadet, J.F.; Calvez, V.; Marcelin, A.G. Quasispecies variant dynamics during emergence of resistance to raltegravir in HIV-1-infected patients. J. Antimicrob. Chemother. 2009, 63, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Ferns, R.B.; Kirk, S.; Bennett, J.; Williams, I.; Edwards, S.; Pillay, D. The dynamics of appearance and disappearance of HIV-1 integrase mutations during and after withdrawal of raltegravir therapy. AIDS 2009, 23, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Canducci, F.; Marinozzi, M.C.; Sampaolo, M.; Boeri, E.; Spagnuolo, V.; Gianotti, N.; Castagna, A.; Paolucci, S.; Baldanti, F.; Lazzarin, A.; Clementi, M. Genotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravir. J. Antimicrob. Chemother. 2010, 65, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Dicker, I.B.; Samanta, H.K.; Li, Z.; Hong, Y.; Tian, Y.; Banville, J.; Remillard, R.R.; Walker M.A.; Langley D.R.; Krystal M. Changes to the HIV LTR and to HIV integrase differentially impact HIV integrase assembly, activity and the binding of strand transfer inhibitors . J. Biol. Chem. 2007, 282, 31186–31196. [Google Scholar] [CrossRef] [PubMed]
- Zahm, J.A.; Bera, S.; Pandey, K.K.; Vora, A.; Stillmock, K.; Hazuda, D.; Grandgenett, D.P. Mechanisms of human immunodeficiency virus type 1 concerted integration related to strand transfer inhibition and drug resistance. Antimicrob. Agents Chemother. 2008, 52, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Passaes, C.B.; Guimarães, M.L.; Fernandez, S.L.C.; Lorete, R.D.S.; Teixeira, S.L.M.; Fernandez, J.C.C.; Morgado, M.G. Lack of primary mutations associated with integrase inhibitors among HIV-1 subtypes B, C, and F circulating in Brazil. J. Acquir. Immune Defic. Syndr. 2009, 51, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Yeh, R.F.; Jain, R.; Palmer, H.R. 49th ICAAC annual meeting: optimization of anti-infective use in the clinical setting. Expert Rev. Anti. Infect. Ther. 2009, 7, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Métifiot, M.; Marchand, C.; Maddali, K.; Pommier, Y. Resistance to Integrase Inhibitors. Viruses 2010, 2, 1347-1366. https://doi.org/10.3390/v2071347
Métifiot M, Marchand C, Maddali K, Pommier Y. Resistance to Integrase Inhibitors. Viruses. 2010; 2(7):1347-1366. https://doi.org/10.3390/v2071347
Chicago/Turabian StyleMétifiot, Mathieu, Christophe Marchand, Kasthuraiah Maddali, and Yves Pommier. 2010. "Resistance to Integrase Inhibitors" Viruses 2, no. 7: 1347-1366. https://doi.org/10.3390/v2071347
APA StyleMétifiot, M., Marchand, C., Maddali, K., & Pommier, Y. (2010). Resistance to Integrase Inhibitors. Viruses, 2(7), 1347-1366. https://doi.org/10.3390/v2071347