The Genomic Diversity and Phylogenetic Relationship in the Family Iridoviridae

Abstract
:
1. Introduction
2. Results and Discussion
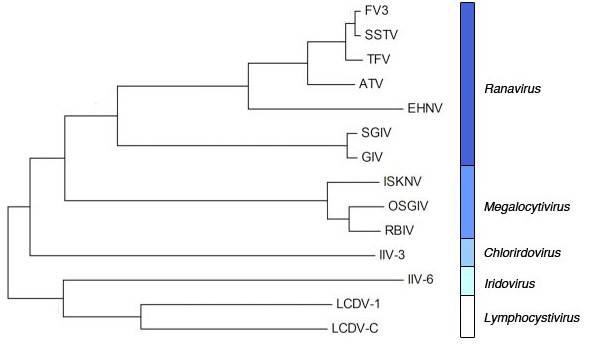
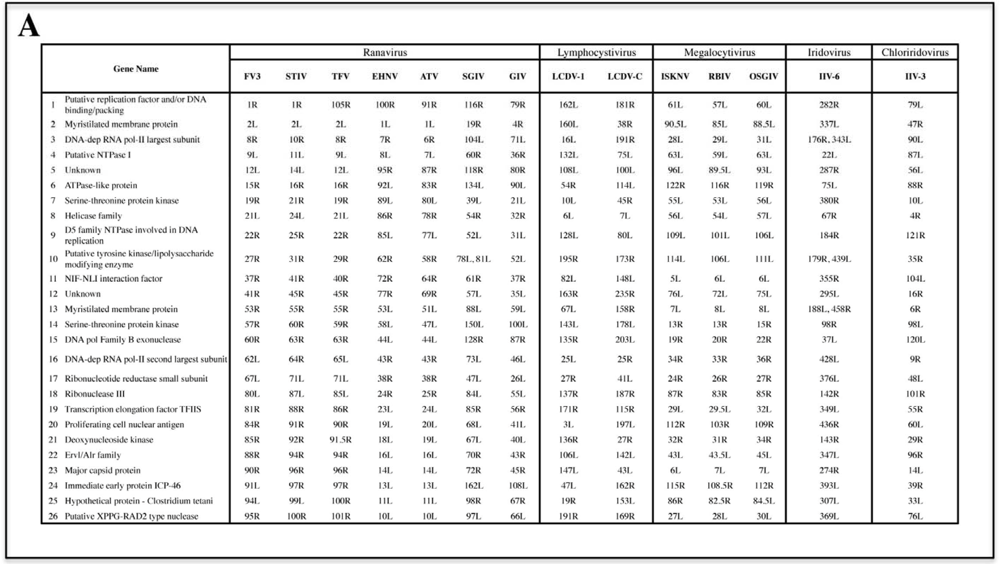
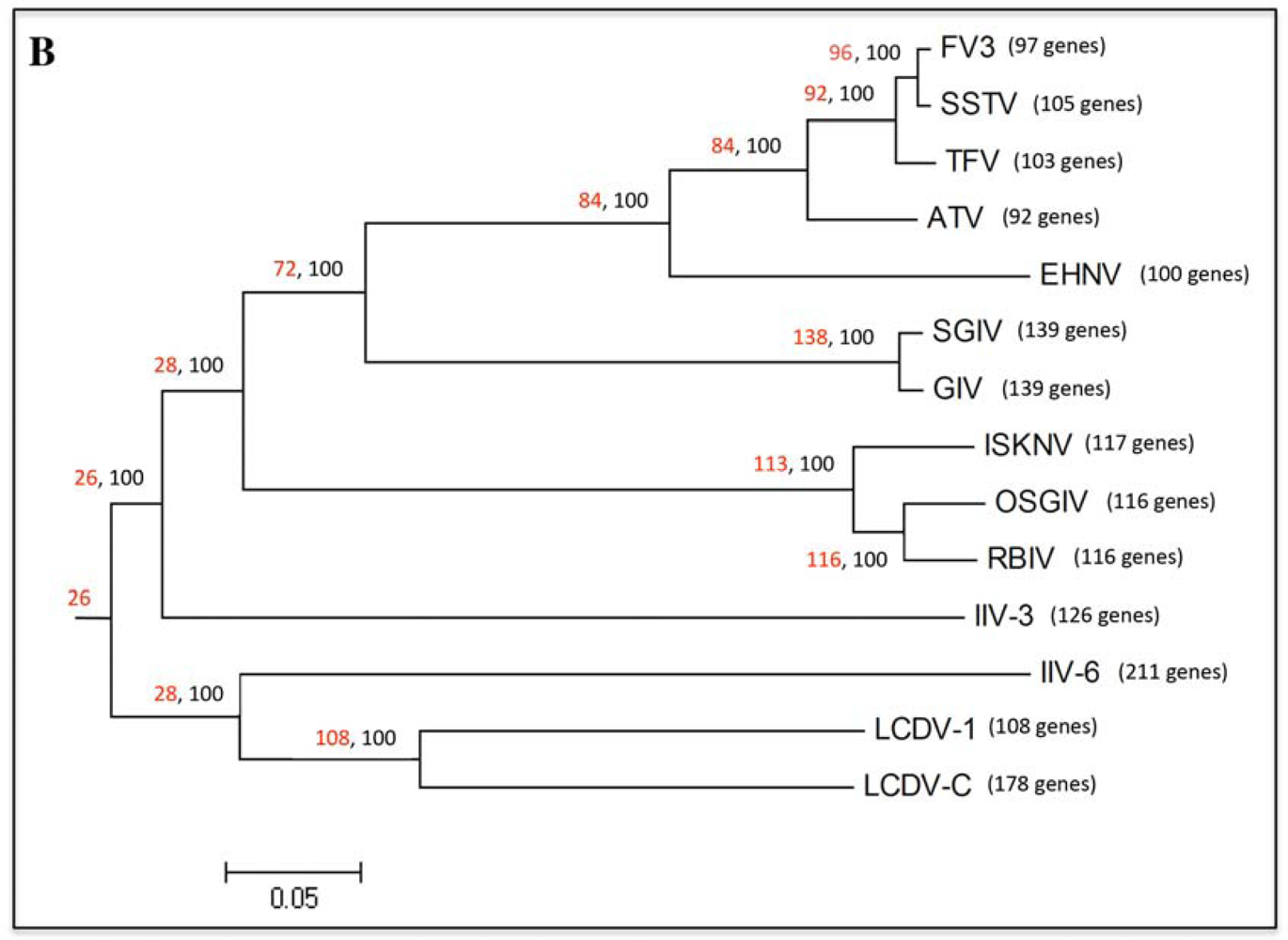
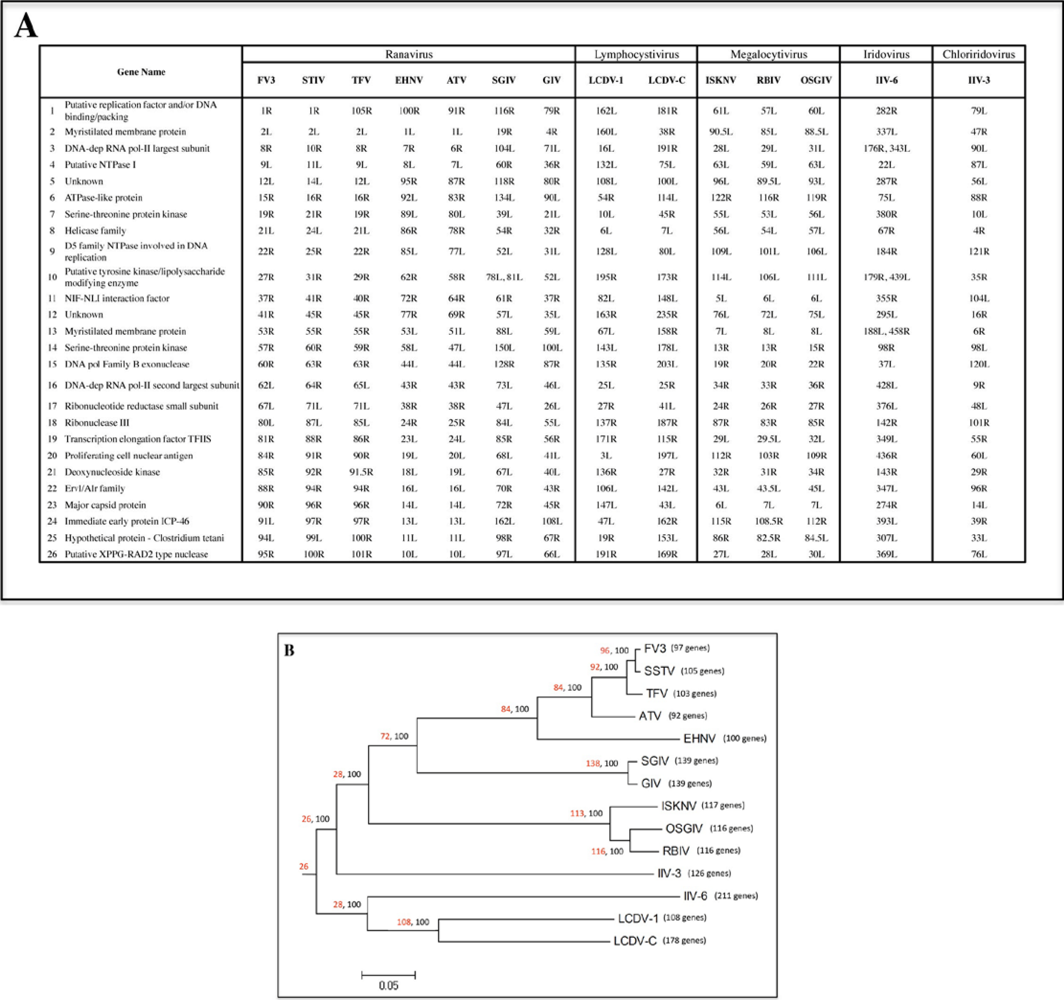
2.1. Phylogenetic analysis
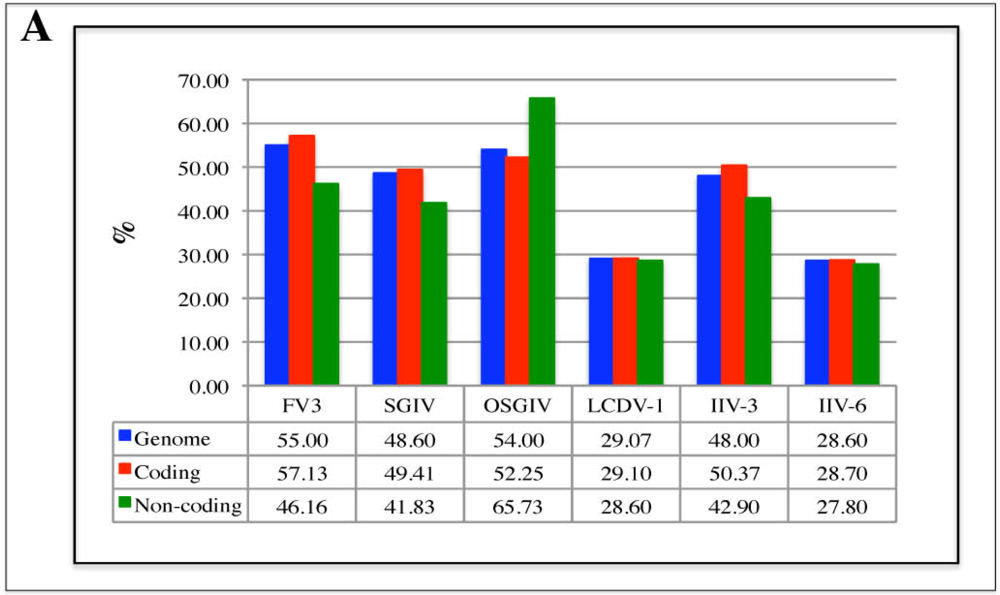
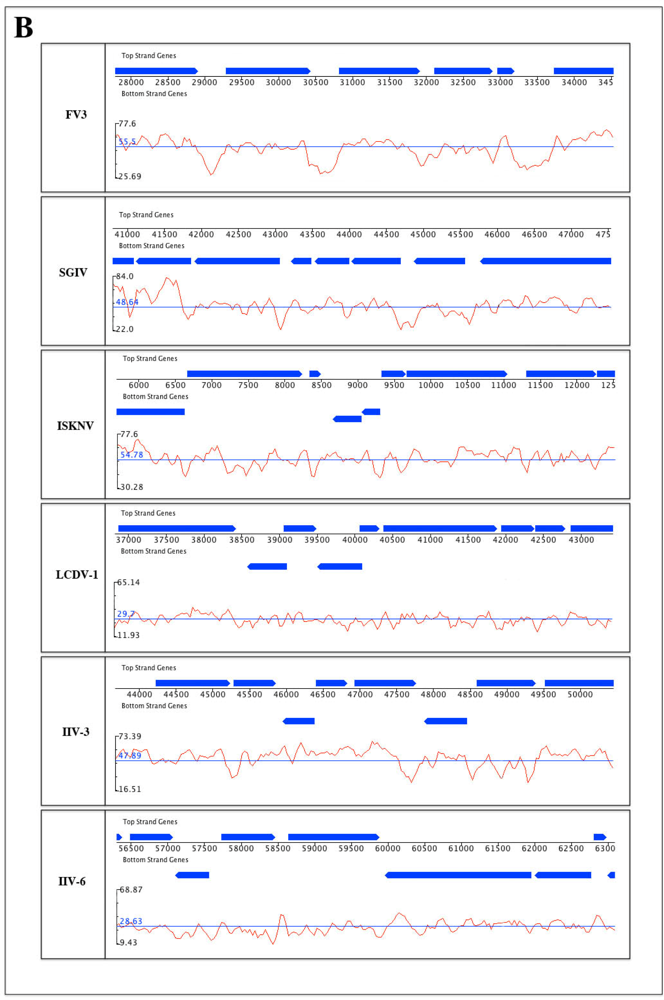
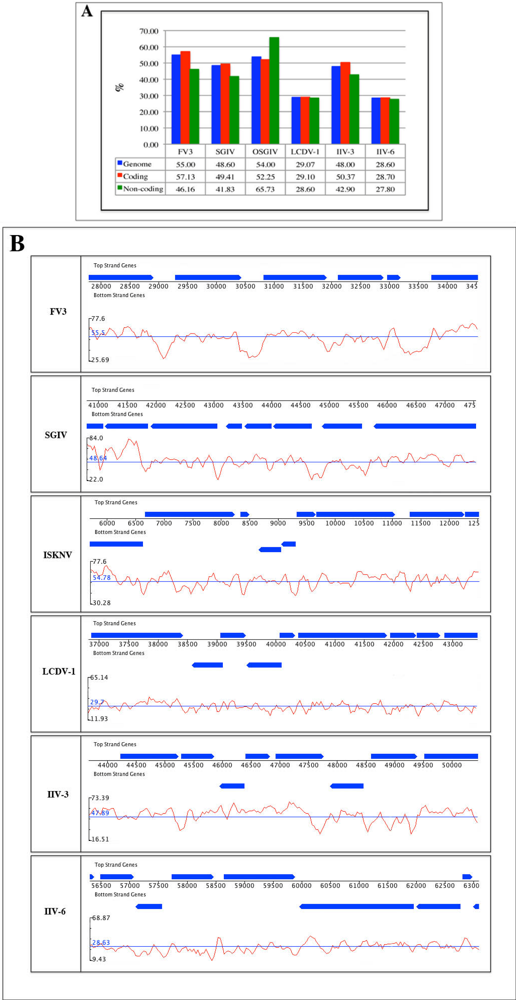
2.2. G/C content
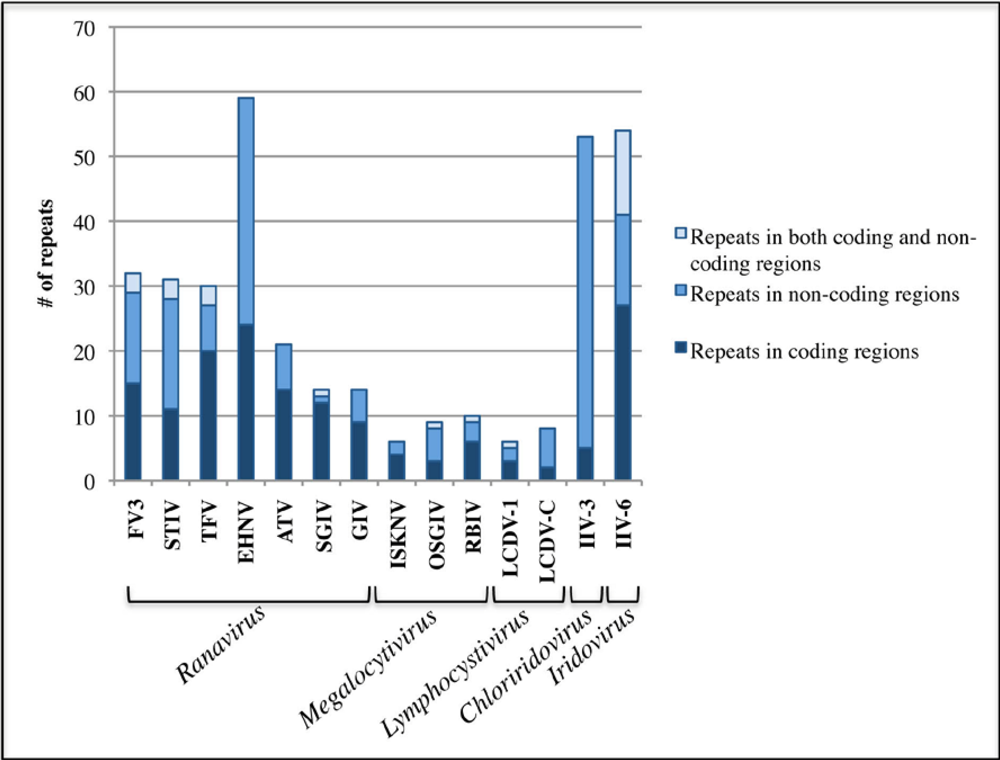
2.3. Iridoviridae repetitive sequences



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |

2.5. Repetitive sequence flanking regions
3. Experimental Section

4. Conclusions
Acknowledgments
References
- He, J.G.; Lu, L.; Deng, M.; He, H.H.; Weng, S.P.; Wang, X.H.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chan, S.M. Sequence analysis of the complete genome of an iridovirus isolated from the tiger frog. Virology 2002, 292, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jakob, N.J.; Muller, K.; Bahr, U.; Darai, G. Analysis of the first complete DNA sequence of an invertebrate iridovirus: coding strategy of the genome of Chilo iridescent virus. Virology 2001, 286, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Chinchar, G.; Darai, G.; Hyatt, A.; Kalmakoff, J.; Seligg, V. Family Iridoviridae. In Virus Taxonomy - 7th report of the international committee on taxonomy of viruses; Regenmortel, M.H.V., Ed.; Academic Press: New York, U.S.A, 2000; pp. 167–182. [Google Scholar]
- Williams, T. The iridoviruses. Adv. Virus Res. 1996, 46, 345–412. [Google Scholar] [PubMed]
- Goorha, R. Frog virus 3 DNA replication occurs in two stages. J. Virol. 1982, 43, 519–528. [Google Scholar] [PubMed]
- Goorha, R.; Murti, G.; Granoff, A.; Tirey, R. Macromolecular synthesis in cells infected by frog virus 3. VIII. The nucleus is a site of frog virus 3 DNA and RNA synthesis. Virology 1978, 84, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Goorha, R.; Murti, K.G. The genome of frog virus 3, an animal DNA virus, is circularly permuted and terminally redundant. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, P.; Soltau, J.B.; Fischer, M.; Reisner, H.; Scholz, J.; Delius, H.; Darai, G. Molecular cloning and physical mapping of the genome of insect iridescent virus type 6: further evidence for circular permutation of the viral genome. Virology 1987, 160, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Delius, H.; Darai, G.; Flugel, R.M. DNA Analysis of Insect Iridescent Virus 6: Evidence for Circular Permutation and Terminal Redundancy. J Virol 1984, 49, 609–614. [Google Scholar] [PubMed]
- Ahne, W.; Schlotfeldt, H.J.; Thomsen, I. Fish viruses: isolation of an icosahedral cytoplasmic deoxyribovirus from sheatfish (Silurus glanis). Zentralbl Veterinarmed B 1989, 36, 333–336. [Google Scholar] [PubMed]
- Hedrick, R.P.; McDowell, T.S. Properties of iridoviruses from ornamental fish. Vet. Res. 1995, 26, 423–427. [Google Scholar] [PubMed]
- Hedrick, R.P.; McDowell, T.S.; Ahne, W.; Torhy, C.; De Kinkelin, P. Properties of three iridovirus-like agents associated with systemic infections of fish. Dis. Aquat. Organ. 1992, 13, 203–209. [Google Scholar] [CrossRef]
- Langdon, J.S.; Humphrey, J.D.; Williams, L.M.; Hyatt, A.D.; Westbury, H.A. First virus isolation from Australian fish: an iridovirus-like pathogen from redfin perch Perca fluviatillis. J. Fish Dis. 1986, 9, 263–268. [Google Scholar] [CrossRef]
- Pozet, F.; Morand, M.; Moussa, A.; Torhy, C.; De Kinkelin, P. Isolation and preliminary characterization of a pathogenic icosahedral deoxyribovirus from the catfish Ictalurus melas. Dis. Aquat. Organ. 1992, 14, 35–42. [Google Scholar] [CrossRef]
- Daszak, P.; Cunningham, A.A.; Hyatt, A.D. Infectious disease and amphibian population declines. Divers. Distrib. 2003, 9, 141–150. [Google Scholar] [CrossRef]
- Jancovich, J.K.; Davidson, E.W.; Parameswaran, N.; Mao, J.; Chinchar, V.G.; Collins, J.P.; Jacobs, B.L.; Storfer, A. Evidence for emergence of an amphibian iridoviral disease because of human-enhanced spread. Mol. Ecol. 2005, 14, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.P.; Storfer, A. Global amphibian declines: sorting the hypotheses. Divers. Distrib. 2003, 9, 89–98. [Google Scholar] [CrossRef]
- Daszak, P.; Berger, L.; Cunningham, A.A.; Hyatt, A.D.; Green, D.E.; Speare, R. Emerging infectious diseases and amphibian population declines. Emerg. Infect. Dis. 1999, 5, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.P.; Brunner, J.L.; Miera, V.; Parris, M.J.; Schock, D.M.; Storfer, A. Ecology and evolution of infectious disease . In Amphibian Conservation; Semlitsch, R., Ed.; Smithsonian Institution Press: Washington, U.S.A., 2003; pp. 137–151. [Google Scholar]
- Tan, W.G.; Barkman, T.J.; Gregory Chinchar, V.; Essani, K. Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 2004, 323, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, X.; Liu, H.; Gong, J.; Ouyang, Z.; Cui, H.; Cao, J.; Zhao, Y.; Wang, X.; Jiang, Y.; Qin, Q. Complete sequence determination of a novel reptile iridovirus isolated from soft-shelled turtle and evolutionary analysis of Iridoviridae. BMC Genomics 2009, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Jancovich, J.K.; Bremont, M.; Touchman, J.W.; Jacobs, B.L. Evidence for multiple recent host species shifts among the Ranaviruses (family Iridoviridae). J. Virol. 2010, 84, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Jancovich, J.K.; Mao, J.; Chinchar, V.G.; Wyatt, C.; Case, S.T.; Kumar, S.; Valente, G.; Subramanian, S.; Davidson, E.W.; Collins, J.P.; Jacobs, B.L. Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mortalities in North America. Virology 2003, 316, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Ting, J.W.; Wu, M.H.; Wu, M.F.; Guo, I.C.; Chang, C.Y. Complete genome sequence of the grouper iridovirus and comparison of genomic organization with those of other iridoviruses. J. Virol. 2005, 79, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Qin, Q.W.; Qiu, J.; Huang, C.H.; Wang, F.; Hew, C.L. Functional genomics analysis of Singapore grouper iridovirus: complete sequence determination and proteomic analysis. J. Virol. 2004, 78, 12576–12590. [Google Scholar] [CrossRef] [PubMed]
- Tidona, C.A.; Darai, G. The complete DNA sequence of lymphocystis disease virus. Virology 1997, 230, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Xiao, F.; Xie, J.; Li, Z.Q.; Gui, J.F. Complete genome sequence of lymphocystis disease virus isolated from China. J. Virol. 2004, 78, 6982–6994. [Google Scholar] [CrossRef] [PubMed]
- He, J.G.; Deng, M.; Weng, S.P.; Li, Z.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chan, S. Complete Genome Analysis of the Mandarin Fish Infectious Spleen and Kidney Necrosis Iridovirus. Virology 2001, 291, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Do, J.W.; Moon, C.H.; Kim, H.J.; Ko, M.S.; Kim, S.B.; Son, J.H.; Kim, J.S.; An, E.J.; Kim, M.K.; Lee, S.K.; Han, M.S.; Cha, S.J.; Park, M.S.; Park, M.A.; Kim, Y.C.; Kim, J.W.; Park, J.W. Complete genomic DNA sequence of rock bream iridovirus. Virology 2004, 325, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Kurita, J.; Nakajima, K.; Hirono, I.; Aoki, T. Complete genome sequencing of red sea bream iridovirus. Fisheries Sci. 2002, 68, 1113–1115. [Google Scholar] [CrossRef]
- Lu, L.; Zhou, S.Y.; Chen, C.; Weng, S.P.; Chan, S.M.; He, J.G. Complete genome sequence analysis of an iridovirus isolated from the orange-spotted grouper, Epinephelus coioides. Virology 2005, 339, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; Becnel, J.J.; Moser, B.A.; Kutish, G.F.; Rock, D.L. Genome of invertebrate iridescent virus type 3 (mosquito iridescent virus). J. Virol. 2006, 80, 8439–8449. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.A.; Jun, S.R.; Sims, G.E.; Kim, S.H. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12826–12831. [Google Scholar] [CrossRef] [PubMed]
- Eaton, H.E.; Metcalf, J.; Penny, E.; Tcherepanov, V.; Upton, C.; Brunetti, C.R. Comparative genomic analysis of the family Iridoviridae: re-annotating and defining the core set of iridovirus genes. Virol. J. 2007, 4, 11. [Google Scholar] [CrossRef]
- Upton, C.; Hogg, D.; Perrin, D.; Boone, M.; Harris, N.L. Viral genome organizer: a system for analyzing complete viral genomes. Virus Res. 2000, 70, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Escudero, R.; Vinuela, E. Structure of African swine fever virus late promoters: requirement of a TATA sequence at the initiation region. J. Virol. 2000, 74, 8176–8182. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxviridae: The viruses and their replication . In Fields Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott Raven: Philadelphia, U.S.A., 1996; pp. 2637–2671. [Google Scholar]
- Nalcacioglu, R.; Marks, H.; Vlak, J.M.; Demirbag, Z.; van Oers, M.M. Promoter analysis of the Chilo iridescent virus DNA polymerase and major capsid protein genes. Virology 2003, 317, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Salas, M.L.; Vinuela, E. Intermediate class of mRNAs in African swine fever virus. J. Virol. 1996, 70, 8584–8589. [Google Scholar] [PubMed]
- Levinson, G.; Gutman, G.A. High frequencies of short frameshifts in poly-CA/TG tandem repeats borne by bacteriophage M13 in Escherichia coli K-12. Nucleic Acids Res. 1987, 15, 5323–5338. [Google Scholar] [CrossRef] [PubMed]
- Wittek, R.; Moss, B. Tandem repeats within the inverted terminal repetition of vaccinia virus DNA. Cell 1980, 21, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, S.; Jacob, R.J.; Roizman, B. Anatomy of herpes simplex virus DNA. II. Size, composition, and arrangement of inverted terminal repetitions . J. Virol. 1975, 15, 1487–1497. [Google Scholar] [PubMed]
- Arrand, J.R.; Roberts, R.J. The nucleotide sequences at the termini of adenovirus-2 DNA. J. Mol. Biol. 1979, 128, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Ko, R.; Okano, K.; Seong, S.I.; Goto, C.; Maeda, S. Sequence analysis of the Xestia c-nigrum granulovirus genome. Virology 1999, 262, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, C.; Goff, S.; Gilboa, E.; Paskind, M.; Mitra, S.W.; Baltimore, D. Structure of a cloned circular Moloney murine leukemia virus DNA molecule containing an inverted segment: implications for retrovirus integration. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 3932–3936. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.; Prolla, T.A.; Liskay, R.M.; Petes, T.D. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993, 365, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [PubMed]
- Hancock, J.M. The contribution of slippage-like processes to genome evolution. J. Mol. Evol. 1995, 41, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Scherer, S.; van Alphen, L.; Verbrugh, H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 1998, 62, 275–293. [Google Scholar] [PubMed]
- Zaia, J.A.; Gallez-Hawkins, G.; Churchill, M.A.; Morton-Blackshere, A.; Pande, H.; Adler, S.P.; Schmidt, G.M.; Forman, S.J. Comparative analysis of human cytomegalovirus a-sequence in multiple clinical isolates by using polymerase chain reaction and restriction fragment length polymorphism assays. J. Clin. Microbiol. 1990, 28, 2602–2607. [Google Scholar] [PubMed]
- Jackson, P.J.; Walthers, E.A.; Kalif, A.S.; Richmond, K.L.; Adair, D.M.; Hill, K.K.; Kuske, C.R.; Andersen, G.L.; Wilson, K.H.; Hugh-Jones, M.; Keim, P. Characterization of the variable-number tandem repeats in vrrA from different Bacillus anthracis isolates. Appl. Environ. Microbiol. 1997, 63, 1400–1405. [Google Scholar] [PubMed]
- Glew, M.D.; Baseggio, N.; Markham, P.F.; Browning, G.F.; Walker, I.D. Expression of the pMGA genes of Mycoplasma gallisepticum is controlled by variation in the GAA trinucleotide repeat lengths within the 5' noncoding regions. Infect. Immun. 1998, 66, 5833–5841. [Google Scholar] [PubMed]
- Field, D.; Wills, C. Abundant microsatellite polymorphism in Saccharomyces cerevisiae, and the different distributions of microsatellites in eight prokaryotes and S. cerevisiae, result from strong mutation pressures and a variety of selective forces. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.J.; Peden, J.F.; Hood, D.W.; Moxon, E.R. Simple sequence repeats in the Helicobacter pylori genome. Mol. Microbiol. 1998, 27, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Gould, A.R.; Zupanovic, Z.; Cunningham, A.A.; Hengstberger, S.; Whittington, R.J.; Kattenbelt, J.; Coupar, B.E. Comparative studies of piscine and amphibian iridoviruses. Arch. Virol. 2000, 145, 301–331. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information Homepage . Available online: http://www.ncbi.nlm.nih.gov/ (accessed 3 April 2010).
- Ehlers, A.; Osborne, J.; Slack, S.; Roper, R.L.; Upton, C. Poxvirus Orthologous Clusters (POCs). Bioinformatics 2002, 18, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Viral Bioinformatics Resource Center Homepage . Available online: www.virology.ca (accessed 26 March 2010).
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Eaton, H.E.; Ring, B.A.; Brunetti, C.R. The Genomic Diversity and Phylogenetic Relationship in the Family Iridoviridae. Viruses 2010, 2, 1458-1475. https://doi.org/10.3390/v2071458
Eaton HE, Ring BA, Brunetti CR. The Genomic Diversity and Phylogenetic Relationship in the Family Iridoviridae. Viruses. 2010; 2(7):1458-1475. https://doi.org/10.3390/v2071458
Chicago/Turabian StyleEaton, Heather E., Brooke A. Ring, and Craig R. Brunetti. 2010. "The Genomic Diversity and Phylogenetic Relationship in the Family Iridoviridae" Viruses 2, no. 7: 1458-1475. https://doi.org/10.3390/v2071458
APA StyleEaton, H. E., Ring, B. A., & Brunetti, C. R. (2010). The Genomic Diversity and Phylogenetic Relationship in the Family Iridoviridae. Viruses, 2(7), 1458-1475. https://doi.org/10.3390/v2071458