Next Generation Sequencing Technologies for Insect Virus Discovery


Abstract
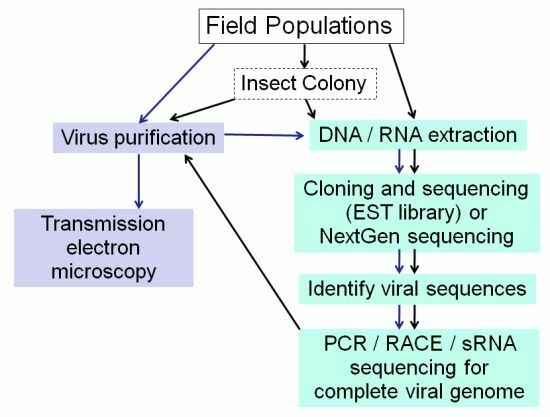
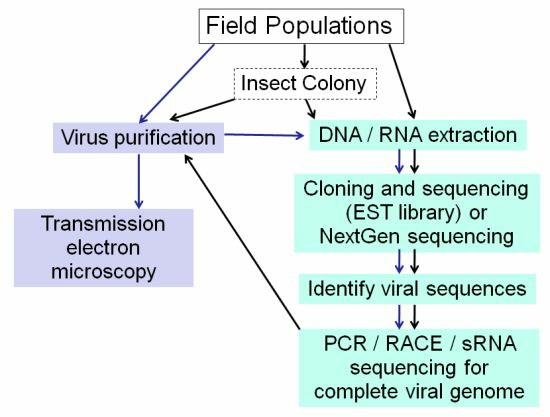
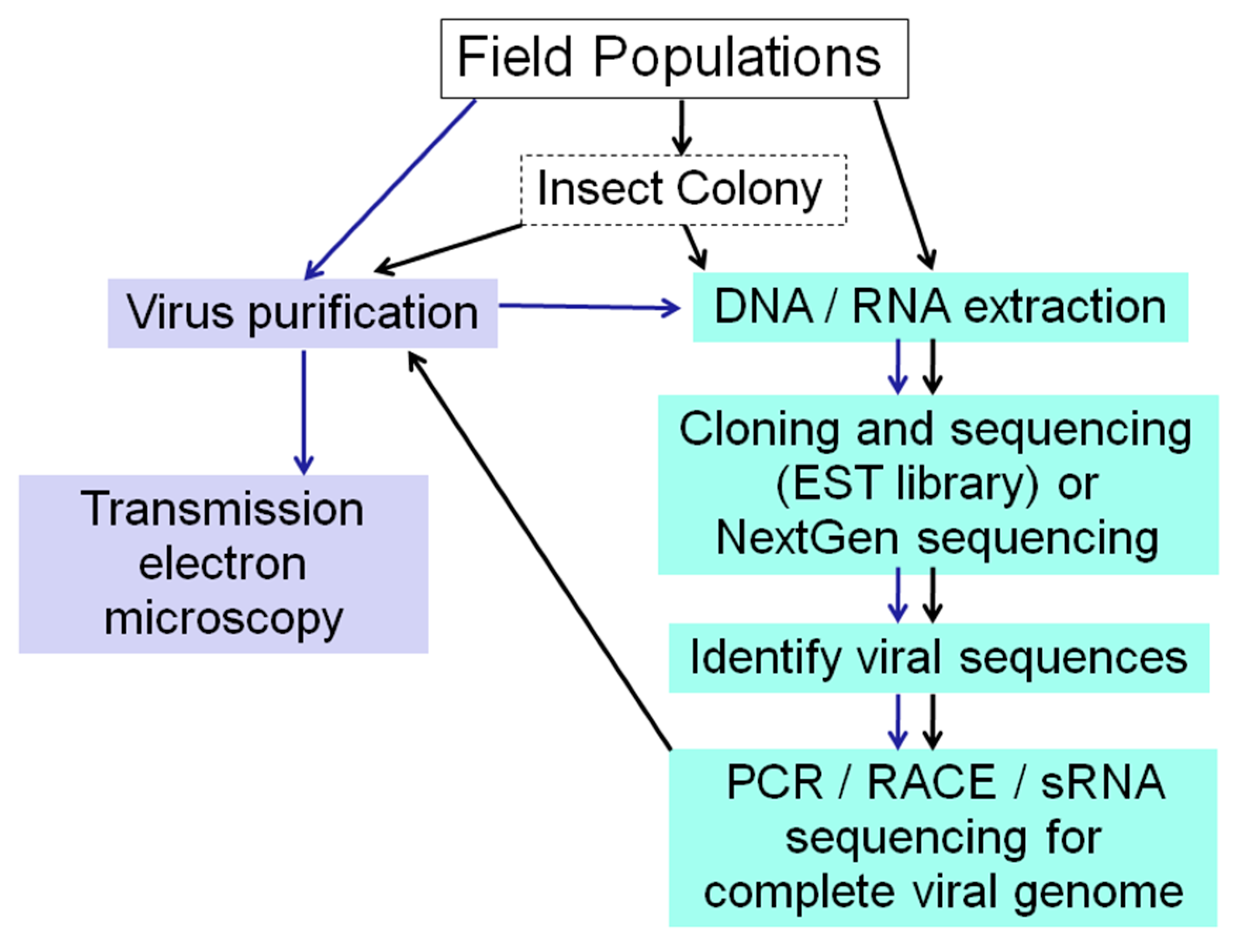
:
1. Introduction
2. Conventional Approaches to Virus Discovery
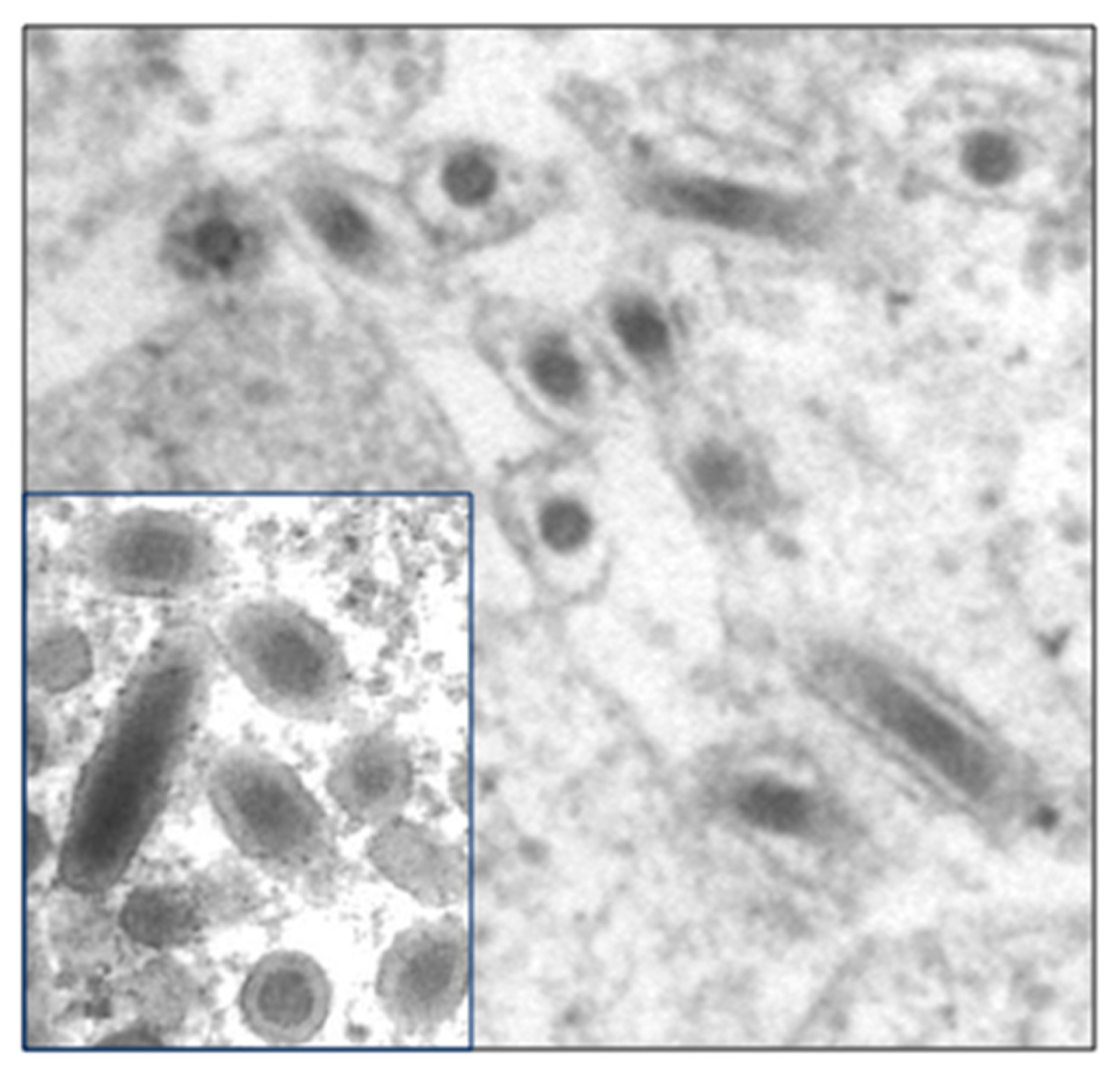
2.1. Electron Microscopy
2.2. Serological Methods
2.3. Standard Molecular Methods
2.4. EST Libraries
2.5. Microarrays
3. Next Generation Sequencing for Virus Discovery
3.1. Sample and Library Preparation
3.2. Bioinformatics Analysis
3.3. Confirmation of NGS-Derived Viral Sequences
4. Discovery of Insect Viruses by NGS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Type | DNA | Transcriptome | Small RNA |
|---|---|---|---|
| Sample and library preparation | Libraries are prepared from DNA isolated from the infected host or from purified viruses. | Libraries are prepared from RNA isolated from the infected host or purified viruses. | Libraries are prepared by isolation of small RNA from host total RNA (~17–30 nt). |
| Treatment of raw data | Base calling, trim adaptors and remove low quality reads. Cluster reads (optional). | ||
| Initial BLAST analysis & assembly | BLAST analysis/mapping followed by assembly of the reads that have significant hits to viral sequences; or assembly of reads and BLAST analysis of the resulting assembled contigs. | Assemble reads followed by BLAST analysis/mapping. | |
| Isolating potential virus sequences | Separate contigs with significant hits (e-value: ≤ 1 × 10−3) to viruses from non-virus hits. | ||
| Re-assemble to generate longer virus contigs | Re-assemble the contigs that hit viral sequences by using various assembly programs (for example software used for Sanger sequence assembly) to generate longer contigs. | ||
| BLAST analysis to identify viruses (known and novel) | BLAST the assembled contigs against non-redundant (nr) databases and virus databases. | ||
| Extend virus genome with overlapping reads with little sequence similarity to known viruses | Identify contigs with hits to viruses [e-value: ≤ 1 × 10−5]. BLAST the viral contigs against the total contig set to search for contigs that overlap viral contigs (but were not identified by BLAST against nr or viral databases). This step is important for identification of novel viral sequences. Assemble virus genomes. | ||
| Generate complete virus genome | Fill the sequence gaps by PCR (RT-PCR, RACE-PCR) and Sanger sequencing. | ||
| Characterize virus | Further characterization of virus (classification, localization, transmission, host range). Refer to polythetic criteria for virus group for parameters needed to facilitate virus classification [54]. | ||
4.1. DNA and Transcriptome Sequencing
4.2. Virus Purification Followed by NGS
4.3. Sequencing of Small RNA
4.4. NGS for the Sequencing of Viral Genomes
4.5. Aphid Virus Discovery using Transcriptome and Small RNA Sequencing
5. Limitations of NGS for Insect Virus Discovery
6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Suttle, C. Crystal ball. The viriosphere: The greatest biological diversity on earth and driver of global processes. Environ. Microbiol. 2005, 7, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Maori, E.; Paldi, N.; Shafir, S.; Kalev, H.; Tsur, E.; Glick, E.; Sela, I. Iapv, a bee-affecting virus associated with colony collapse disorder can be silenced by dsRNA ingestion. Insect Mol. Biol. 2009, 18, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Moscardi, F. Assessment of the application of baculoviruses for control of lepidoptera. Annu. Rev. Entomol. 1999, 44, 257–289. [Google Scholar] [CrossRef]
- Gray, S.M.; Banerjee, N. Mechanisms of arthropod transmission of plant and animal viruses. Microbiol. Mol. Biol. Rev. 1999, 63, 128–148. [Google Scholar] [CrossRef] [PubMed]
- van Oers, M.M. Opportunities and challenges for the baculovirus expression system. J. Invertebr. Pathol. 2011, 107, S3–S15. [Google Scholar] [CrossRef]
- Bonning, B.C. Insect Viruses: Biotechnological Applications; Elsevier: San Diego, CA, USA, 2006; Volume 68, p. 532. [Google Scholar]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Microbiol. 2011, 9, 99–108. [Google Scholar] [CrossRef]
- Schmidt, O.; Theopold, U.; Strand, M. Innate immunity and its evasion and suppression by hymenopteran endoparasitoids. Bioessays 2001, 23, 344–351. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Keane, G.; Naish, N.; Evered, C.; Winstanley, D. Densovirus induces winged morphs in asexual clones of the rosy apple aphid, dysaphis plantaginea. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8465–8470. [Google Scholar] [CrossRef]
- Oliver, K.M.; Degnan, P.H.; Burke, G.R.; Moran, N.A. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu. Rev. Entomol. 2010, 55, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Degnan, P.H.; Hunter, M.S.; Moran, N.A. Bacteriophages encode factors required for protection in a symbiotic mutualism. Science 2009, 325, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Lilly, K.A.; Turell, M.J.; Willner, K.M.; Butani, A.; Nolan, N.M.; Lentz, S.M.; Akmal, A.; Mateczun, A.; Brahmbhatt, T.N.; Sozhamannan, S.; et al. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl. Trop. Dis. 2010, 4, e878. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.B.; Patte, C.P.; Sinisterra, X.H.; Achor, D.S.; Funk, C.J.; Polston, J.E. Discovering new insect viruses: Whitefly iridovirus (homoptera: Aleyrodidae: Bemisia tabaci). J. Invertebr. Pathol. 2001, 78, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.J.; Hunter, W.B.; Achor, D.S. Replication of insect iridescent virus 6 in a whitefly cell line. J. Invertebr. Pathol. 2001, 77, 144–146. [Google Scholar] [CrossRef]
- Zirkel, F.; Kurth, A.; Quan, P.L.; Briese, T.; Ellerbrok, H.; Pauli, G.; Leendertz, F.H.; Lipkin, W.I.; Ziebuhr, J.; Drosten, C.; et al. An insect nidovirus emerging from a primary tropical rainforest. MBio 2011, 2, e00077-11. [Google Scholar] [CrossRef]
- Bai, H.; Wang, Y.; Li, X.; Mao, H.; Li, Y.; Han, S.; Shi, Z.; Chen, X. Isolation and characterization of a novel alphanodavirus. Virol. J. 2011, 8, 311. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Miller, S.E. Modern uses of electron microscopy for detection of viruses. Clin. Microbiol. Rev. 2009, 22, 552–563. [Google Scholar] [CrossRef]
- Garrett, R.G.; O’Loughlin, G.T. Broccoli necrotic yellows virus in cauliflower and in the aphid, brevicoryne brassicae l. Virology 1977, 76, 653–663. [Google Scholar] [CrossRef]
- Parrish, W.B.; Briggs, J.D. Morphological identification of virus like particles in the corn leaf aphid, rhopalosiphum maidis (fitch.). J. Invertebr. Pathol. 1966, 8, 122–123. [Google Scholar] [CrossRef]
- Peters, D. The purification of virus like particles from the aphid myzus persicae. Virology 1965, 26, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.Y.; Huang, C.Y.; Wang, C.H.; Chiang, H.C.; Lo, C.F. Pathogenicity of a baculovirus infection causing white spot syndrome in cultured penaeid shrimp in taiwan. Dis. Aquat. Org. 1995, 23, 165–173. [Google Scholar] [CrossRef]
- Hayat, M.A.; Miller, S.E. Negative Staining: Applications and Methods; McGraw-Hill: New York, NY, USA, 1990. [Google Scholar]
- Almeida, J.D.; Waterson, A.D. Some implications of a morphologically oriented classification of viruses. Arch. Gesamte Virusforsch 1970, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.F.; Stanley, W.M. A study by means of the electron microscope of the reaction between tobacco mosaic virus and its antiserum. J. Biol. Chem. 1941, 139, 339–344. [Google Scholar] [CrossRef]
- Boyapalle, S.; Beckett, R.J.; Pal, N.; Miller, W.A.; Bonning, B.C. Infectious genomic RNA of rhopalosiphum padi virus transcribed in vitro from a full-length cdna clone. Virology 2008, 375, 401–411. [Google Scholar] [CrossRef]
- Valles, S.M.; Strong, C.A.; Hunter, W.B.; Dang, P.M.; Pereira, R.M.; Oi, D.H.; Williams, D.F. Expressed sequence tags from the red imported fire ant, solenopsis invicta: Annotation and utilization for discovery of viruses. J. Invertebr. Pathol. 2008, 99, 74–81. [Google Scholar] [CrossRef]
- Hunnicutt, L.E.; Hunter, W.B.; Cave, R.D.; Powell, C.A.; Mozoruk, J.J. Genome sequence and molecular characterization of homalodisca coagulata virus-1, a novel virus discovered in the glassy-winged sharpshooter (hemiptera: Cicadellidae). Virology 2006, 350, 67–78. [Google Scholar] [CrossRef]
- Hunter, W.B.; Katsar, C.S.; Chaparro, J.X. Molecular analysis of capsid protein of homalodisca coagulata virus-1, a new leafhopper-infecting virus from the glassy-winged sharpshooter, homalodisca coagulata. J. Insect Sci. 2006, 6, 1–10. [Google Scholar] [CrossRef]
- Katsar, C.S.; Hunter, W.B.; Sinisterra, X.H. Phytoreovirus-like sequences isolated from salivary glands of the glassy-winged sharpshooter homolodisca vitripennis (hemiptera: Cicadellidae) Fla. Entomol. 2007, 90, 196–203. [Google Scholar]
- Oliveira, D.C.; Hunter, W.B.; Ng, J.; Desjardins, C.A.; Dang, P.M.; Werren, J.H. Data mining cdnas reveals three new single stranded RNA viruses in nasonia (hymenoptera: Pteromalidae). Insect Mol. Biol. 2010, 19, 99–107. [Google Scholar] [CrossRef]
- Wang, D.; Coscoy, L.; Zylberberg, M.; Avila, P.C.; Boushey, H.A.; Ganem, D.; DeRisi, J.L. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15687–15692. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Urisman, A.; Liu, Y.T.; Springer, M.; Ksiazek, T.G.; Erdman, D.D.; Mardis, E.R.; Hickenbotham, M.; Magrini, V.; Eldred, J.; et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003, 1, E2. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; Flenniken, M.L.; Engel, J.C.; Ruby, J.G.; Ganem, D.; Andino, R.; Derisi, J.L. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, nosema, and crithidia. PLoS One 2011, 6, e20656. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef]
- Hall, N. Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol. 2007, 210, 1518–1525. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.A.; Porreca, G.J.; Church, G.M. Overview of DNA sequencing strategies. Curr. Protoc. Mol. Biol. 2008. Chapter 7, Unit 7.1. [Google Scholar] [CrossRef]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.L.; Briese, T.; Hornig, M.; Geiser, D.M.; et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef]
- Ng, T.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS One 2011, 6, e20579. [Google Scholar] [CrossRef]
- Rusk, N. Focus on next-generation sequencing data analysis. Forward. Nat. Methods 2009, 6, S1. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Cutadapt. Available online: http://code.google.com/p/cutadapt/ (accessed on 18 June 2011).
- Altschul, S.F.; Gish, W.; Miller, W.M.; Eyers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Plaskon, N.E.; Adelman, Z.N.; Myles, K.M. Accurate strand-specific quantification of viral RNA. PLoS One 2009, 4, e7468. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- van Vliet, A.H. Next generation sequencing of microbial transcriptomes: Challenges and opportunities. FEMS Microbiol. Lett. 2010, 302, 1–7. [Google Scholar] [CrossRef]
- Wang, L.F. Discovering novel zoonotic viruses. N S W Public Health Bull. 2011, 22, 113–117. [Google Scholar] [CrossRef]
- Studholme, D.J.; Glover, R.H.; Boonham, N. Application of high-throughput DNA sequencing in phytopathology. Annu. Rev. Phytopathol. 2011, 49, 87–105. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. Metagenomic analysis of coastal RNA virus communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef]
- Updates on programs available can be found at the following sites: http://seqanswers.com/wiki/Software/list; http://www.oxfordjournals.org/our_journals/bioinformatics/nextgenerationsequencing.html; https://wiki.nbic.nl/index.php/NGS_Tools (accessed on 1 August 2011).
- ICTV International committee on taxonomy of viruses—Approved virus orders, families and genera. Available online: http://ictvonline.org/virusTaxonomy.asp?version=2009 (accessed on 1 August 2011).
- van Engelsdorp, D.; Meixner, M.D. A historical review of managed honey bee populations in europe and the united states and the factors that may affect them. J. Invertebr. Pathol. 2010, 103, S80–S95. [Google Scholar] [CrossRef]
- Genersch, E.; Evans, J.D.; Fries, I. Honey bee disease overview. J. Invertebr. Pathol. 2010, 103, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Djikeng, A.; Kuzmickas, R.; Anderson, N.G.; Spiro, D.J. Metagenomic analysis of RNA viruses in a fresh water lake. PLoS One 2009, 4, e7264. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1606–1611. [Google Scholar] [CrossRef]
- Liu, S.; Vijayendran, D.; Bonning, B.C. Iowa State University: Ames, IA, USA, Unpublished work. 2011.
- Chen, Y.F.; Gao, F.; Ye, X.Q.; Wei, S.J.; Shi, M.; Zheng, H.J.; Chen, X.X. Deep sequencing of cotesia vestalis bracovirus reveals the complexity of a polydnavirus genome. Virology 2011, 414, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Abd-Alla, A.M.; Cousserans, F.; Parker, A.G.; Jehle, J.A.; Parker, N.J.; Vlak, J.M.; Robinson, A.S.; Bergoin, M. Genome analysis of a glossina pallidipes salivary gland hypertrophy virus reveals a novel, large, double-stranded circular DNA virus. J. Virol. 2008, 82, 4595–4611. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. High diversity of unknown picorna-like viruses in the sea. Nature 2003, 424, 1054–1057. [Google Scholar] [CrossRef]
- Ding, S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef]
- van Mierlo, J.T.; van Cleef, K.W.; van Rij, R.P. Defense and counterdefense in the rnai-based antiviral immune system in insects. Methods Mol. Biol. 2011, 721, 3–22. [Google Scholar]
- Zambon, R.A.; Vakharia, V.N.; Wu, L.P. Rnai is an antiviral immune response against a dsRNA virus in drosophila melanogaster. Cell Microbiol. 2006, 8, 880–889. [Google Scholar] [CrossRef]
- Li, H.; Li, W.X.; Ding, S.W. Induction and suppression of RNA silencing by an animal virus. Science 2002, 296, 1319–1321. [Google Scholar] [CrossRef]
- Galiana-Arnoux, D.; Dostert, C.; Schneemann, A.; Hoffmann, J.A.; Imler, J.L. Essential function in vivo for dicer-2 in host defense against RNA viruses in drosophila. Nat. Immunol. 2006, 7, 590–597. [Google Scholar] [CrossRef] [PubMed]
- van Rij, R.P.; Saleh, M.C.; Berry, B.; Foo, C.; Houk, A.; Antoniewski, C.; Andino, R. The RNA silencing endonuclease argonaute 2 mediates specific antiviral immunity in drosophila melanogaster. Genes Dev. 2006, 20, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Aliyari, R.; Li, W.X.; Li, H.W.; Kim, K.; Carthew, R.; Atkinson, P.; Ding, S.W. RNA interference directs innate immunity against viruses in adult drosophila. Science 2006, 312, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Sabin, L.R.; Zhou, R.; Gruber, J.J.; Lukinova, N.; Bambina, S.; Berman, A.; Lau, C.K.; Thompson, C.B.; Cherry, S. Ars2 regulates both miRNA- and siRNA- dependent silencing and suppresses RNA virus infection in drosophila. Cell 2009, 138, 340–351. [Google Scholar] [CrossRef]
- Chotkowski, H.L.; Ciota, A.T.; Jia, Y.; Puig-Basagoiti, F.; Kramer, L.D.; Shi, P.Y.; Glaser, R.L. West nile virus infection of drosophila melanogaster induces a protective rnai response. Virology 2008, 377, 197–206. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, S.; Navarro, B.; Gisel, A.; Pena, L.; Navarro, L.; Moreno, P.; Di Serio, F.; Flores, R. Citrus tristeza virus infection induces the accumulation of viral small RNAs (21–24-nt) mapping preferentially at the 3′-terminal region of the genomic RNA and affects the host small RNA profile. Plant Mol. Biol. 2011, 75, 607–619. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Wang, Y.; Sheng, G.; Juranek, S.; Tuschl, T.; Patel, D.J. Structure of the guide-strand-containing argonaute silencing complex. Nature 2008, 456, 209–213. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Tettelin, H.; Radune, D.; Kasif, S.; Khouri, H.; Salzberg, S.L. Optimized multiplex pcr: Efficiently closing a whole-genome shotgun sequencing project. Genomics 1999, 62, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. Abyss: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- van Munster, M.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.; Clerivet, A.; van der Wilk, F. Sequence analysis and genomic organization of aphid lethal paralysis virus: A new member of the family dicistroviridae. J. Gen. Virol. 2002, 83, 3131–3138. [Google Scholar] [CrossRef]
- DArcy, C.J.; Burnett, P.A.; Hewings, A.D. Detection, biological effects and transmission of a virus of the aphid rhopalosiphum padi. Virology 1981, 114, 268–272. [Google Scholar] [CrossRef]
- van Munster, M.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.F.; Reinbold, C.; Brault, V.; Clerivet, A.; van der Wilk, F. Characterization of a new densovirus infecting the green peach aphid myzus persicae. J. Invertebr. Pathol. 2003, 84, 6–14. [Google Scholar] [CrossRef]
- van Munster, M.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.F.; Reinbold, C.; Brault, V.; Clerivet, A.; van der Wilk, F. A new virus infecting myzus persicae has a genome organization similar to the species of the genus densovirus. J. Gen. Virol. 2003, 84, 165–172. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Sparks, W.O.; Bartholomay, L.; Bonning, B.C. Insect immunity to viruses. In Insect Immunology; Beckage, N.E., Ed.; Academic Press: San Diego, CA, USA, 2008; pp. 209–242. [Google Scholar]
- Clem, R.J. Baculoviruses and apoptosis: A diversity of genes and responses. Curr. Drug Targets 2007, 8, 1069–1074. [Google Scholar] [CrossRef]
- Nayak, A.; Berry, B.; Tassetto, M.; Kunitomi, M.; Acevedo, A.; Deng, C.; Krutchinsky, A.; Gross, J.; Antoniewski, C.; Andino, R. Cricket paralysis virus antagonizes argonaute 2 to modulate antiviral defense in drosophila. Nat. Struct. Mol. Biol. 2010, 17, 547–554. [Google Scholar] [CrossRef]
- Berry, B.; Deddouche, S.; Kirschner, D.; Imler, J.L.; Antoniewski, C. Viral suppressors of RNA silencing hinder exogenous and endogenous small RNA pathways in drosophila. PLoS One 2009, 4, e5866. [Google Scholar] [CrossRef]
- Clarke, T.E.; Clem, R.J. Insect defenses against virus infection: The role of apoptosis. Int. Rev. Immunol. 2003, 22, 401–424. [Google Scholar] [CrossRef] [PubMed]
- Gerardo, N.M.; Altincicek, B.; Anselme, C.; Atamian, H.; Barribeau, S.M.; de Vos, M.; Duncan, E.J.; Evans, J.D.; Gabaldon, T.; Ghanim, M.; et al. Immunity and other defenses in pea aphids, acyrthosiphon pisum. Genome Biol. 2010, 11, R21. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.A.; Bogaert, T.; Clinton, W.; Heck, G.R.; Feldmann, P.; Ilagan, O.; Johnson, S.; Plaetinck, G.; Munyikwa, T.; Pleau, M.; et al. Control of coleopteran insect pests through RNA interference. Nat. Biotechnol. 2007, 25, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Terenius, O.; Papanicolaou, A.; Garbutt, J.S.; Eleftherianos, I.; Huvenne, H.; Kanginakudru, S.; Albrechtsen, M.; An, C.; Aymeric, J.L.; Barthel, A.; et al. RNA interference in lepidoptera: An overview of successful and unsuccessful studies and implications for experimental design. J. Insect Physiol. 2011, 57, 231–245. [Google Scholar] [CrossRef] [PubMed]



| Platform | Roche 454/GS FLX + | Illumina GAII | Life Technologies / SOLiD 5500xl system | |
|---|---|---|---|---|
| GAII | HiSeq 2000 | |||
| Library | Fragment / emulsion PCR | Fragment / polony | Fragment / emulsion PCR | |
| Sequencing Principal | Pyrosequencing | Sequencing by synthesis | Sequencing by ligation | |
| Read length (base) | 700–1000 | 150 | 100 | 75 |
| Gb per run | 0.7 | 95 | 600 | 300 |
| Pros | Long reads improve mapping in repetitive regions, fast run time | Currently the most widely used platform in the field | Two-base encoding provides inherent error correction | |
| Cons | High reagent cost, high error rate in homopolymer repeats | Low multiplexing capability of samples | Long run time | |
| Examples of biological applications | Bacterial and insect genome de novo assemblies, medium scale (<3 Mb) exome capture, virus discovery in metagenomics | Variant discovery by whole—genome resequencing or whole—exome capture, virus discovery and gene discovery in metagenomics | Variant discovery by whole—genome resequencing or whole—exome capture, gene discovery in metagenomics | |
| Virus | Origin | Reference |
|---|---|---|
| Birnaviridae (dsRNA) | ||
| Drosophila X virus (DXV) | D. melanogaster cell line (S2-GMR) | [58] |
| Drosophila birnavirus (DBV)* | D. melanogaster cell line (S2-GMR) | [58] |
| Totiviridae (dsRNA) | ||
| Drosophila totivirus (DTV)* | D. melanogaster cell line (S2-GMR) | [58] |
| Dicistroviridae (+ssRNA) | ||
| Drosophila C virus (DCV) | D. melanogaster ovary somatic cell line | [58] |
| Black queen cell virus (BQCV) | Apis mellifera | [41] |
| Kashmir bee virus (KBV) | Apis mellifera | [41] |
| Acute bee paralysis virus (ABPV) | Apis mellifera | [41] |
| Isreali acute paralysis virus (IAPV) | Apis mellifera | [41,57] |
| Aphid lethal paralysis virus-AP (ALPV-AP) | Acyrthosiphon pisum | [59] |
| ALPV-AG | Aphis glycines | [59] |
| ALPV-Brookings strain (ALPV-Brookings)* | Apis mellifera | [35] |
| Big Sioux river virus (BSRV)* | Apis mellifera | [35] |
| Nodaviridae (+ssRNA) | ||
| American nodavirus (ANV)* | D. melanogaster cell line (S2-GMR) | [58] |
| Mosquito nodavirus (MNV)* | Aedes aegypti-Liverpool strain | [58] |
| Nidovirales (+ssRNA) | ||
| Cavally virus (CAVV)* | Mosquito heads (multiple species) | [17] |
| Tetraviridae (+ssRNA) | ||
| Drosophila tetravirus (DTrV)*1 | D. melanogaster cell lines, S2-GMR & Kc | [58] |
| Togaviridae (+ssRNA) | ||
| Sindbis virus (SINV) | Aedes aegypti-Liverpool strain | [58] |
| Picornaviridae (+ssRNA) | ||
| Deformed wing virus (DWV) | Apis mellifera | [41] |
| Sacbrood virus (SBV) | Apis mellifera | [41] |
| Polydnaviridae | ||
| Costesia vestalis bracovirus (CvBV) | Costesia vestalis | [60] |
| Parvoviridae (ssDNA) | ||
| Myzus persicae densovirus (MpDNV) | Myzus persicae | [59] |
| Unclassified | ||
| Noravirus (+ssRNA)* | D. melanogaster ovary cell line | [58] |
| Chronic bee paralysis virus (CBPV; +ssRNA) | Apis mellifera | [41] |
| Glossina pallidipes salivary gland hypertrophy virus (GpSGHV;dsDNA) | Glossina pallidipes salivary glands | [61] |
| Lake Sinai Virus 1 (LSV1;+ssRNA)* | Apis mellifera | [35] |
| Lake Sinai Virus 2 (LSV2;+ssRNA)* | Apis mellifera | [35] |
| Aphis glycines virus (AGV;+ssRNA)* | Aphis glycines | [59] |
| Others | ||
| Many DNA viruses (known and novel) from animal, plant, insect | Various species of female mosquitoes | [42] |
| Many known DNA and RNA viruses | Apis mellifera | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Vijayendran, D.; Bonning, B.C. Next Generation Sequencing Technologies for Insect Virus Discovery. Viruses 2011, 3, 1849-1869. https://doi.org/10.3390/v3101849
Liu S, Vijayendran D, Bonning BC. Next Generation Sequencing Technologies for Insect Virus Discovery. Viruses. 2011; 3(10):1849-1869. https://doi.org/10.3390/v3101849
Chicago/Turabian StyleLiu, Sijun, Diveena Vijayendran, and Bryony C. Bonning. 2011. "Next Generation Sequencing Technologies for Insect Virus Discovery" Viruses 3, no. 10: 1849-1869. https://doi.org/10.3390/v3101849
APA StyleLiu, S., Vijayendran, D., & Bonning, B. C. (2011). Next Generation Sequencing Technologies for Insect Virus Discovery. Viruses, 3(10), 1849-1869. https://doi.org/10.3390/v3101849