What Has the Study of the K3 and K5 Viral Ubiquitin E3 Ligases Taught Us about Ubiquitin-Mediated Receptor Regulation?

Abstract
:1. Introduction
2. K3 and K5 Modulate Host Cell Surface Receptors
3. Bioinformatic Clues to K3 and K5’s Mechanism of Action
4. The RING-CH Domains of mK3, K3 and K5 Direct Ubiquitination
5. mK3 is an ERAD E3 Ligase
6. Endocytosis Induced by K3 and K5
7. K3 and K5 Expose New Amino Acid Targets of Ubiquitination
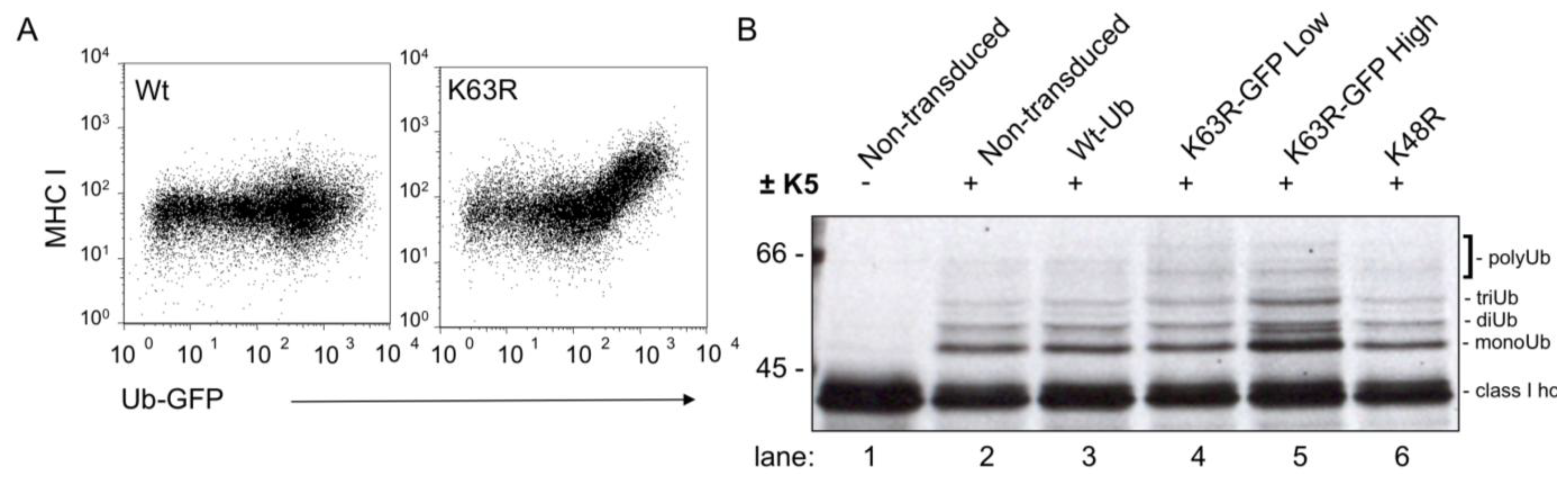
8. K3 and K5 Recruit Consecutive E2s to Polyubiquitinate MHC I Molecules via Lys63 Linkages for Endolysosomal Degradation
9. Internalization of MHC I by K5 Requires Mixed Lys11 and Lys63 Linked Polyubiquitin Chains on a Single Lysine Acceptor Residue
10. Do Other Polyubiquitin Chains Substitute for Lys63 in MHC I Downregulation?
11. Cellular Co-Factors Required for Endocytosis and Degradation of Polyubiquitinated Substrates
12. The K3 and K5 Viral E3 Ligases were Appropriated from the MARCH Family of Human E3 Ligases
13. Conclusions
Acknowledgements
References and Notes
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Ferber, S.; Ciechanover, A. Transfer RNA is required for conjugation of ubiquitin to selective substrates of the ubiquitin- and ATP-dependent proteolytic system. J. Biol. Chem. 1986, 261, 3128–3134. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 2010, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Markson, G.; Kiel, C.; Hyde, R.; Brown, S.; Charalabous, P.; Bremm, A.; Semple, J.; Woodsmith, J.; Duley, S.; Salehi-Ashtiani, K.; Vidal, M.; Komander, D.; Serrano, L.; Lehner, P.; Sanderson, C.M. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 2009, 19, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Bremm, A.; Freund, S.M.; Komander, D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol. 2010, 17, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Lehner, P.J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; Efstathiou, S.; Doherty, P.C.; Lehner, P.J. Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8455–8460. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8051–8056. [Google Scholar] [CrossRef]
- Ishido, S.; Choi, J.K.; Lee, B.S.; Wang, C.; DeMaria, M.; Johnson, R.P.; Cohen, G.B.; Jung, J.U. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 2000, 13, 365–374. [Google Scholar] [CrossRef]
- Haque, M.; Ueda, K.; Nakano, K.; Hirata, Y.; Parravicini, C.; Corbellino, M.; Yamanishi, K. Major histocompatibility complex class I molecules are down-regulated at the cell surface by the K5 protein encoded by Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. J. Gen. Virol. 2001, 82, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7–2 and modulates T cell costimulation. J. Clin. Invest. 2001, 107, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; Stevenson, P.G. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 2001, 15, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.; Zong, J.; Ciufo, D.; Guo, H.G.; Reitz, M.S.; Hayward, G.S. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J. Virol. 1997, 71, 1963–1974. [Google Scholar] [CrossRef]
- Dodd, R.B.; Allen, M.D.; Brown, S.E.; Sanderson, C.M.; Duncan, L.M.; Lehner, P.J.; Bycroft, M.; Read, R.J. Solution structure of the Kaposi's sarcoma-associated herpesvirus K3 N-terminal domain reveals a Novel E2-binding C4HC3-type RING domain. J. Biol. Chem. 2004, 279, 53840–53847. [Google Scholar] [CrossRef]
- Bartee, E.; Mansouri, M.; Hovey Nerenberg, B.T.; Gouveia, K.; Fruh, K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef]
- Lehner, P.J.; Hoer, S.; Dodd, R.; Duncan, L.M. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol. Rev. 2005, 207, 112–125. [Google Scholar] [CrossRef]
- Hewitt, E.W.; Duncan, L.; Mufti, D.; Baker, J.; Stevenson, P.G.; Lehner, P.J. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 2002, 21, 2418–2429. [Google Scholar] [CrossRef]
- Coscoy, L.; Sanchez, D.J.; Ganem, D. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 2001, 155, 1265–1273. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Lencer, W.; Hansen, T.H. The viral E3 ubiquitin ligase mK3 uses the Derlin/p97 endoplasmic reticulum-associated degradation pathway to mediate down-regulation of major histocompatibility complex class I proteins. J. Biol. Chem. 2006, 281, 8636–8644. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Harris, M.R.; Lybarger, L.; Kimpler, L.A.; Myers, N.B.; Virgin, H.W.t.; Hansen, T.H. Physical association of the K3 protein of gamma-2 herpesvirus 68 with major histocompatibility complex class I molecules with impaired peptide and beta(2)-microglobulin assembly. J. Virol. 2002, 76, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; de Lima, B.D.; Lehner, P.J.; Stevenson, P.G. Viral degradation of the MHC class I peptide loading complex. Immunity 2004, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; May, J.S.; Stevenson, P.G. The murine gamma-herpesvirus-68 MK3 protein causes TAP degradation independent of MHC class I heavy chain degradation. Eur. J. Immunol. 2005, 35, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Douglas, J.; Rose, P.P.; Gouveia, K.; Thomas, G.; Means, R.E.; Moses, A.V.; Fruh, K. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 2006, 108, 1932–1940. [Google Scholar] [CrossRef]
- Sanchez, D.J.; Gumperz, J.E.; Ganem, D. Regulation of CD1d expression and function by a herpesvirus infection. J. Clin. Invest. 2005, 115, 1369–1378. [Google Scholar] [CrossRef]
- Manes, T.D.; Hoer, S.; Muller, W.A.; Lehner, P.J.; Pober, J.S. Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. J. Immunol. 2010, 184, 5186–5192. [Google Scholar] [CrossRef]
- Li, Q.; Means, R.; Lang, S.; Jung, J.U. Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol. 2007, 81, 2117–2127. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Boyle, L.H.; Boname, J.M.; Lehner, P.J.; Trowsdale, J. Ubiquitination of lysine-331 by Kaposi's sarcoma-associated herpesvirus protein K5 targets HFE for lysosomal degradation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16240–16245. [Google Scholar] [CrossRef]
- Mansouri, M.; Rose, P.P.; Moses, A.V.; Fruh, K. Remodeling of endothelial adherens junctions by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 9615–9628. [Google Scholar] [CrossRef]
- Thomas, M.; Boname, J.M.; Field, S.; Nejentsev, S.; Salio, M.; Cerundolo, V.; Wills, M.; Lehner, P.J. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1656–1661. [Google Scholar] [CrossRef]
- Bartee, E.; McCormack, A.; Fruh, K. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; Hines, J.; Gustin, J.; Moses, A.V.; Fruh, K. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009, 83, 9672–9681. [Google Scholar] [CrossRef] [PubMed]
- Pardieu, C.; Vigan, R.; Wilson, S.J.; Calvi, A.; Zang, T.; Bieniasz, P.; Kellam, P.; Towers, G.J.; Neil, S.J. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 2010, 6, e1000843. [Google Scholar] [CrossRef]
- Durrington, H.J.; Upton, P.D.; Hoer, S.; Boname, J.; Dunmore, B.J.; Yang, J.; Crilley, T.K.; Butler, L.M.; Blackbourn, D.J.; Nash, G.B.; Lehner, P.J.; Morrell, N.W. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J. Biol. Chem. 2010, 285, 37641–37649. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; Thomas, M.; Stagg, H.R.; Xu, P.; Peng, J.; Lehner, P.J. Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 2010, 11, 210–220. [Google Scholar] [CrossRef]
- Cadwell, K.; Coscoy, L. The specificities of Kaposi’s sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J. Virol. 2008, 82, 4184–4189. [Google Scholar] [CrossRef]
- Cadwell, K.; Coscoy, L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 2005, 309, 127–130. [Google Scholar] [CrossRef]
- Wang, X.; Herr, R.A.; Chua, W.J.; Lybarger, L.; Wiertz, E.J.; Hansen, T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef]
- Vosper, J.M.; McDowell, G.S.; Hindley, C.J.; Fiore-Heriche, C.S.; Kucerova, R.; Horan, I.; Philpott, A. Ubiquitylation on canonical and non-canonical sites targets the transcription factor neurogenin for ubiquitin-mediated proteolysis. J. Biol. Chem. 2009, 284, 15458–15468. [Google Scholar] [CrossRef]
- Ishikura, S.; Weissman, A.M.; Bonifacino, J.S. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J. Biol. Chem. 2010, 285, 23916–23924. [Google Scholar] [CrossRef]
- Galan, J.M.; Haguenauer-Tsapis, R. Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J. 1997, 16, 5847–5854. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Di Fiore, P.P.; Dikic, I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 2003, 28, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef]
- Traub, L.M.; Lukacs, G.L. Decoding ubiquitin sorting signals for clathrin-dependent endocytosis by CLASPs. J. Cell Sci. 2007, 120, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Barriere, H.; Nemes, C.; Lechardeur, D.; Khan-Mohammad, M.; Fruh, K.; Lukacs, G.L. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in Mammalian cells. Traffic 2006, 7, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.M.; Piper, S.; Dodd, R.B.; Saville, M.K.; Sanderson, C.M.; Luzio, J.P.; Lehner, P.J. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J. 2006, 25, 1635–1645. [Google Scholar] [CrossRef]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Geetha, T.; Jiang, J.; Wooten, M.W. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 2005, 20, 301–312. [Google Scholar] [CrossRef]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef]
- Varadan, R.; Assfalg, M.; Haririnia, A.; Raasi, S.; Pickart, C.; Fushman, D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J. Biol. Chem. 2004, 279, 7055–7063. [Google Scholar] [CrossRef]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef]
- Duncan, L.M.; Nathan, J.A.; Lehner, P.J. Stabilization of an E3 ligase-E2-ubiquitin complex increases cell surface MHC class I expression. J. Immunol. 2010, 184, 6978–6985. [Google Scholar] [CrossRef]
- Kirkpatrick, D.S.; Hathaway, N.A.; Hanna, J.; Elsasser, S.; Rush, J.; Finley, D.; King, R.W.; Gygi, S.P. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 2006, 8, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Yamanaka, Y.; Ishikawa, A.; Aoki-Kawasumi, M.; Mito-Yoshida, M.; Ohmura-Hoshino, M.; Matsuki, Y.; Kajikawa, M.; Hirano, H.; Ishido, S. Contribution of Lysine 11-linked Ubiquitination to MIR2-mediated Major Histocompatibility Complex Class I Internalization. J. Biol. Chem. 2010, 285, 35311–35319. [Google Scholar] [CrossRef]
- Bowers, K.; Piper, S.C.; Edeling, M.A.; Gray, S.R.; Owen, D.J.; Lehner, P.J.; Luzio, J.P. Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J. Biol. Chem. 2006, 281, 5094–5105. [Google Scholar] [CrossRef]
- Collin, N.; Guerin, J.L.; Drexler, I.; Blanie, S.; Gelfi, J.; Boullier, S.; Foucras, G.; Sutter, G.; Messud-Petit, F. The poxviral scrapin MV-LAP requires a myxoma viral infection context to efficiently downregulate MHC-I molecules. Virology 2005, 343, 171–178. [Google Scholar] [CrossRef] [PubMed]
- De Gassart, A.; Camosseto, V.; Thibodeau, J.; Ceppi, M.; Catalan, N.; Pierre, P.; Gatti, E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3491–3496. [Google Scholar] [CrossRef]
- Ishido, S.; Matsuki, Y.; Goto, E.; Kajikawa, M.; Ohmura-Hoshino, M. MARCH-I: A new regulator of dendritic cell function. Mol. Cells 2010, 29, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Walseng, E.; Furuta, K.; Bosch, B.; Weih, K.A.; Matsuki, Y.; Bakke, O.; Ishido, S.; Roche, P.A. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 170, 20465–20470. [Google Scholar] [CrossRef]
- Hoer, S.; Smith, L.; Lehner, P.J. MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett. 2007, 581, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hor, S.; Ziv, T.; Admon, A.; Lehner, P.J. Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantitation identify new substrates for the MARCH9 transmembrane E3 ligase. Mol. Cell. Proteomics 2009, 8, 1959–1971. [Google Scholar] [CrossRef]
- Nice, T.J.; Deng, W.; Coscoy, L.; Raulet, D.H. Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J. Immunol. 2010, 185, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Herr, R.A.; Rabelink, M.; Hoeben, R.C.; Wiertz, E.J.; Hansen, T.H. Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J. Cell Biol. 2009, 187, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Fukuda, H.; Kato, A.; Hirose, S. MARCH-II is a syntaxin-6-binding protein involved in endosomal trafficking. Mol. Biol. Cell 2005, 16, 1696–1710. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Nakamura, N.; Hirose, S. MARCH-III Is a novel component of endosomes with properties similar to those of MARCH-II. J. Biochem. 2006, 139, 137–145. [Google Scholar] [CrossRef]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef]
- Hassink, G.; Kikkert, M.; van Voorden, S.; Lee, S.J.; Spaapen, R.; van Laar, T.; Coleman, C.S.; Bartee, E.; Fruh, K.; Chau, V.; Wiertz, E. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef]
- Nathan, J.A.; Sengupta, S.; Wood, S.A.; Admon, A.; Markson, G.; Sanderson, C.; Lehner, P.J. The ubiquitin E3 ligase MARCH7 is differentially regulated by the deubiquitylating enzymes USP7 and USP9X. Traffic 2008, 9, 1130–1145. [Google Scholar] [CrossRef]
- Morokuma, Y.; Nakamura, N.; Kato, A.; Notoya, M.; Yamamoto, Y.; Sakai, Y.; Fukuda, H.; Yamashina, S.; Hirata, Y.; Hirose, S. MARCH-XI, a novel transmembrane ubiquitin ligase implicated in ubiquitin-dependent protein sorting in developing spermatids. J. Biol. Chem. 2007, 282, 24806–24815. [Google Scholar] [CrossRef]


{kind=link}
| E3 ligases | substrates | 1° Ub acceptor | E2s | linkage | DUBs |
|---|---|---|---|---|---|
| K3 | MHC I (HLA-A, HLA-B, HLA-C, HLA-E), CD1d, PECAM, IFN-γR1 | Lys or Cys 10-15 amino acids from the membrane | UBE2D (UbcH5) and UBE2N (Ubc13) | Lys63 | |
| K5 | MHC I (HLA-A, HLA-B, weakly HLA-C), HFE, CD1d, MIC-A, MIC-B, AICL, ICAM-1, PECAM, ALCAM, VE-cadherin, B7-2, IFN-γR1, Syntaxin-4, BMPRII, BST-2/tetherin | membrane proximal Lys or Cys | UBE2D (UbcH5) and UBE2N (Ubc13) | Lys63, Lys11 | |
| mK3 | MHC I, tapasin, TAP | Lys, Ser or Thr | Ube2J2 [64] | Lys48 | |
| MARCH 1 | MHC II | Lys | |||
| MARCH 2 | Transferrin receptor, B7-2, DLG1, binds syntaxin 6 [65] | ||||
| MARCH 3 | binds syntaxin 6 [66] | ||||
| MARCH 4 | MHC I, CD4, Mult1 (mice) [63] | Lys | Lys63 | ||
| MARCH 5 | Drp1 [67] | ||||
| MARCH 6 (TEB4) | ERAD ligase [68] | ||||
| MARCH 7 | unknown | USP7, USP9X [69] | |||
| MARCH 8 | MHC II, B7-2, Transferrin receptor | Lys | |||
| MARCH 9 | MHC I, ICAM-1, CD4, FCγRIIB, CD150, HLA-DQ, PTPRJ, ILT-2, Mult1 (mice) | Lys | |||
| MARCH 10 | unknown | ||||
| MARCH 11 | binds Veli-3 [70] |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boname, J.M.; Lehner, P.J. What Has the Study of the K3 and K5 Viral Ubiquitin E3 Ligases Taught Us about Ubiquitin-Mediated Receptor Regulation? Viruses 2011, 3, 118-131. https://doi.org/10.3390/v3020118
Boname JM, Lehner PJ. What Has the Study of the K3 and K5 Viral Ubiquitin E3 Ligases Taught Us about Ubiquitin-Mediated Receptor Regulation? Viruses. 2011; 3(2):118-131. https://doi.org/10.3390/v3020118
Chicago/Turabian StyleBoname, Jessica M., and Paul J. Lehner. 2011. "What Has the Study of the K3 and K5 Viral Ubiquitin E3 Ligases Taught Us about Ubiquitin-Mediated Receptor Regulation?" Viruses 3, no. 2: 118-131. https://doi.org/10.3390/v3020118
APA StyleBoname, J. M., & Lehner, P. J. (2011). What Has the Study of the K3 and K5 Viral Ubiquitin E3 Ligases Taught Us about Ubiquitin-Mediated Receptor Regulation? Viruses, 3(2), 118-131. https://doi.org/10.3390/v3020118