Characterization of a Full-Length Endogenous Beta-Retrovirus, EqERV-Beta1, in the Genome of the Horse (Equus caballus)


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
3. Results
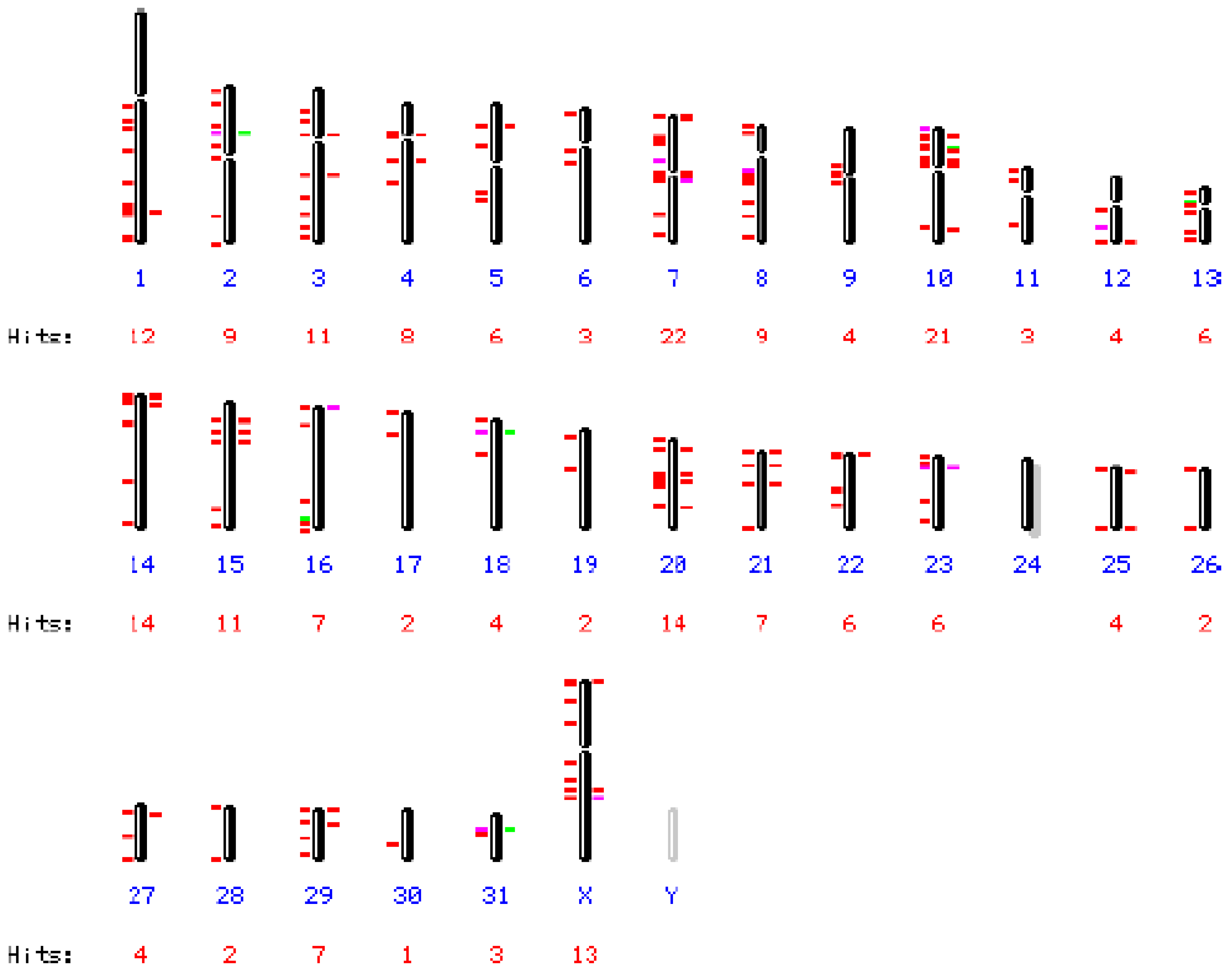
3.1. Detection of Endogenous Retrovirus pol Fragments in the Horse Genome
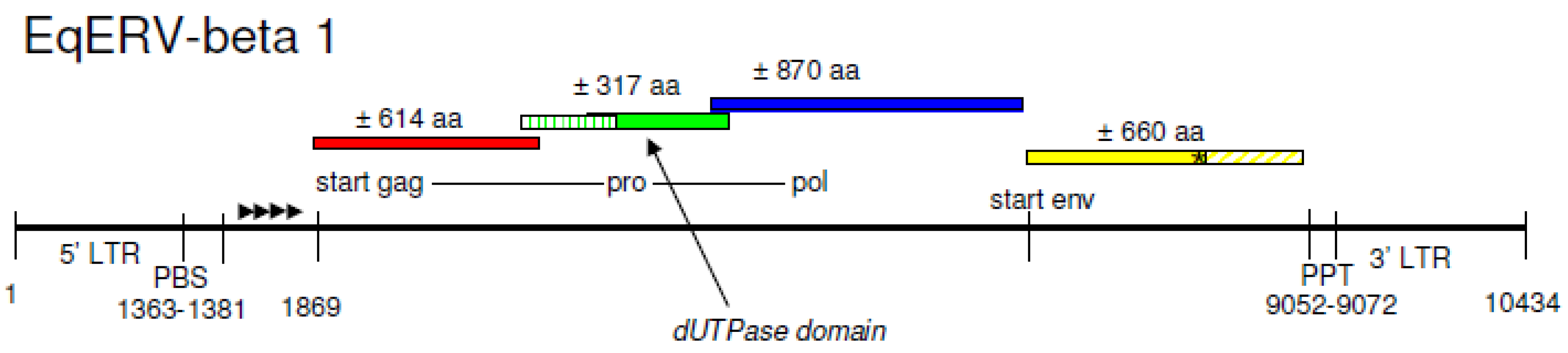
3.2. A Complete Beta-Retrovirus Genome Is Integrated on Horse Chromosome 5
3.3. Elements Related to EqERV-Beta1 in the Horse Genome
3.4. The LTR of the Horse Endogenous Retrovirus EqERV-Beta1
3.5. Analysis of the Reading Frames of the Horse Endogenous Retrovirus EqERV-Beta1
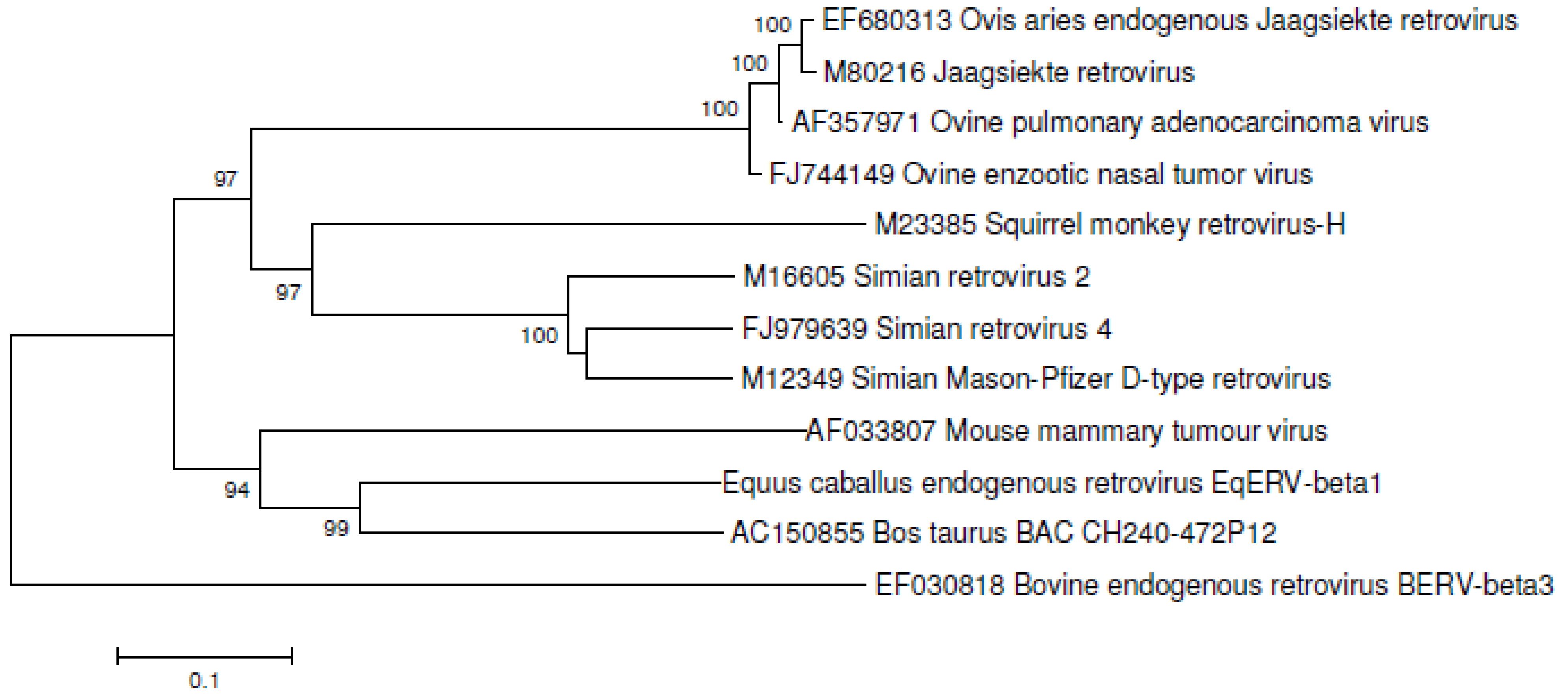
3.6. Phylogenetic Analysis of EqERV-Beta1
4. Discussion and Conclusions
Acknowledgements
References and Notes
- Horie, M.; Honda, T.; Suzuki, Y.; Kobayashi, Y.; Daito, T.; Oshida, T.; Ikuta, K.; Jern, P.; Gojobori, T.; Coffin, J.M.; Tomonaga, K. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 2010, 463, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Belyi, V.A.; Levine, A.J.; Skalka, A.M. Unexpected inheritance: multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PLoS. Pathog. 2010, 6, e1001030. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.J.; Leach, R.W.; Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC. Evol. Biol. 2010, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Taya, K.; Sashihara, J.; Kurahashi, H.; Amo, K.; Miyagawa, H.; Kondo, K.; Okada, S.; Yamanishi, K. Human herpesvirus 6 (HHV-6) is transmitted from parent to child in an integrated form and characterization of cases with chromosomally integrated HHV-6 DNA. J. Med. Virol. 2004, 73, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. The discovery of endogenous retroviruses. Retrovirology. 2006, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Sperber, G.O.; Blomberg, J. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology. 2005, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J.; Katzourakis, A.; Tristem, M.; Pybus, O.G.; Winters, M.; Shafer, R.W. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 20362–20367. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, W.; Abrantes, J; Esteves, P.J. Sharing of endogenous lentiviral gene fragments among leporid lineages separated for more than 12 million years. J. Virol. 2009, 83, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Keckesova, Z.; Ylinen, L.M.; Towers, G.J.; Gifford, R.J.; Katzourakis, A. Identification of a RELIK orthologue in the European hare (Lepus europaeus) reveals a minimum age of 12 million years for the lagomorph lentiviruses. Virology 2009, 384, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.D.; Lee, F.; Capelli, C.; DeSalle, R.; Tikhonov, A.; Marx, P.A.; MacPhee, R.D. Evolution of endogenous retrovirus-like elements of the woolly mammoth (Mammuthus primigenius) and its relatives. Mol. Biol. Evol. 2001, 18, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Benit, L.; Lallemand, J.B.; Casella, J.F.; Philippe, H.; Heidmann, T. ERV-L elements: a family of endogenous retrovirus-like elements active throughout the evolution of mammals. J. Virol. 1999, 73, 3301–3308. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Horse (Equus caballus) Genome Browser Gateway of the Genome Bioinformatics Group of UC Santa Cruz. Available online: http://genome.ucsc.edu/cgi-bin/hgGateway?db=equCab2 (accessed on 4 January 2011).
- Horse Genome Resources, NCBI. Available online: http://www.ncbi.nlm.nih.gov/projects/genome/guide/horse/ (accessed on 8 November 2010).
- NCBI Basic Local Alignment Search Tool BLAST. Available online: http://blast.ncbi.nlm.nih.gov/ (accessed on 8 November 2010).
- NCBI nucleotide database. Available online: www.ncbi.nlm.nih.gov/nucleotide/ (accessed on 8 November 2010).
- BioEdit Sequence Alignment Editor, Version 7.0.9. Available online: www.mbio.ncsu.edu/BioEdit/bioedit.html (accessed on 14 October 2010).
- MEGA 4 software package. Available online: www.megasoftware.net (accessed on 14 October 2010).
- Marquet, R.; Isel, C.; Ehresmann, C.; Ehresmann, B. tRNAs as primer of reverse transcriptases. Biochimie 1995, 77, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Shimotohno, K.; Mizutani, S.; Temin, H.M. Sequence of retrovirus provirus resembles that of bacterial transposable elements. Nature 1980, 285, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.M.; Abecasis, G.R.; Altshuler, D.L.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar]
- Benachenhou, F.; Jern, P.; Oja, M.; Sperber, G.; Blikstad, V.; Somervuo, P.; Kaski, S.; Blomberg, J. Evolutionary conservation of orthoretroviral long terminal repeats (LTRs) and ab initio detection of single LTRs in genomic data. PLoS. ONE 2009, 4, e5179. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Meese, E.U. Presence of dUTPase in the various human endogenous retrovirus K (HERV-K) families. J. Mol. Evol. 2003, 57, 642–649. [Google Scholar] [CrossRef] [PubMed]
- van der Kuyl, A.C.; Mang, R.; Dekker, J.T.; Goudsmit, J. Complete nucleotide sequence of simian endogenous type D retrovirus with intact genome organization: evidence for ancestry to simian retrovirus and baboon endogenous virus. J. Virol. 1997, 71, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; Weese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, 37, D205–D210. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Kuyl, A.C. Characterization of a Full-Length Endogenous Beta-Retrovirus, EqERV-Beta1, in the Genome of the Horse (Equus caballus). Viruses 2011, 3, 620-628. https://doi.org/10.3390/v3060620
Van der Kuyl AC. Characterization of a Full-Length Endogenous Beta-Retrovirus, EqERV-Beta1, in the Genome of the Horse (Equus caballus). Viruses. 2011; 3(6):620-628. https://doi.org/10.3390/v3060620
Chicago/Turabian StyleVan der Kuyl, Antoinette C. 2011. "Characterization of a Full-Length Endogenous Beta-Retrovirus, EqERV-Beta1, in the Genome of the Horse (Equus caballus)" Viruses 3, no. 6: 620-628. https://doi.org/10.3390/v3060620
APA StyleVan der Kuyl, A. C. (2011). Characterization of a Full-Length Endogenous Beta-Retrovirus, EqERV-Beta1, in the Genome of the Horse (Equus caballus). Viruses, 3(6), 620-628. https://doi.org/10.3390/v3060620