Molecular and Cellular Aspects of Rhabdovirus Entry

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Basic Biochemical Properties of the Rhabdovirus Glycoprotein
3. Rhabdovirus Receptors
4. Fusion Properties of Rhabdoviruses
5. Structural Studies on VSV G
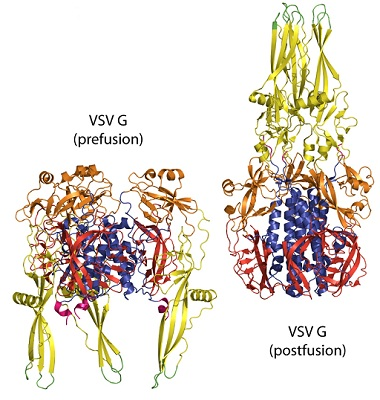
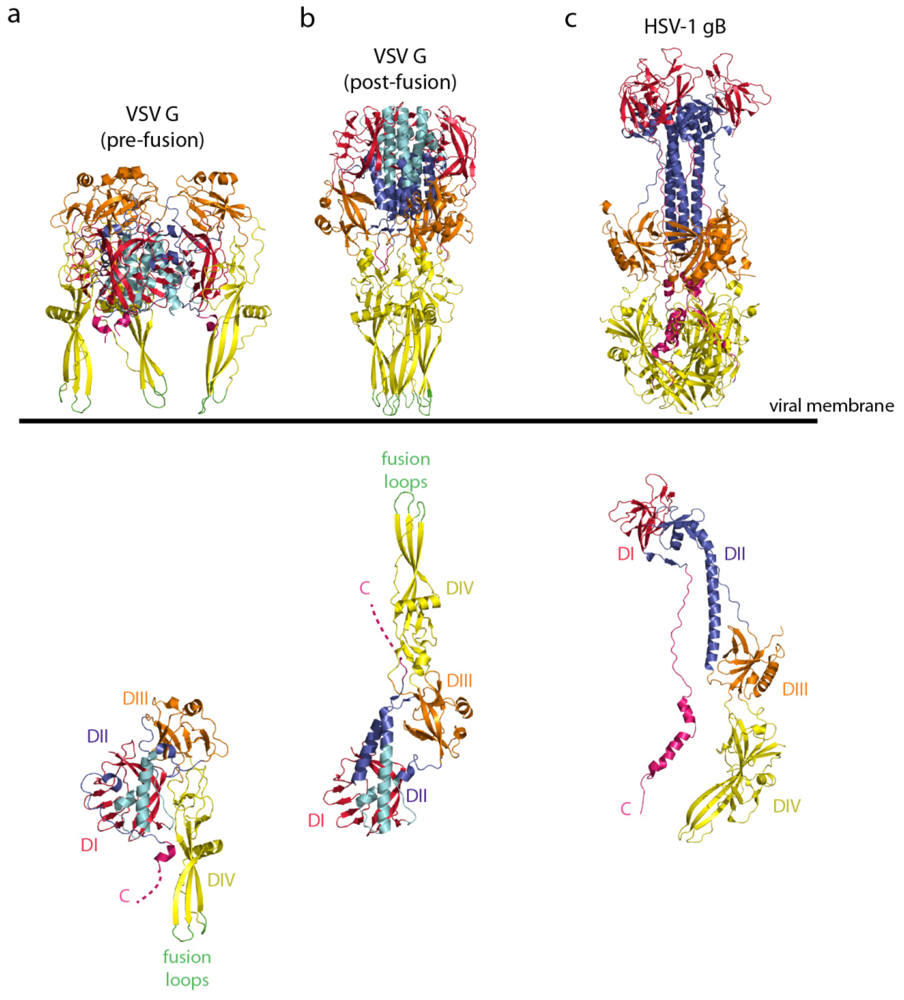
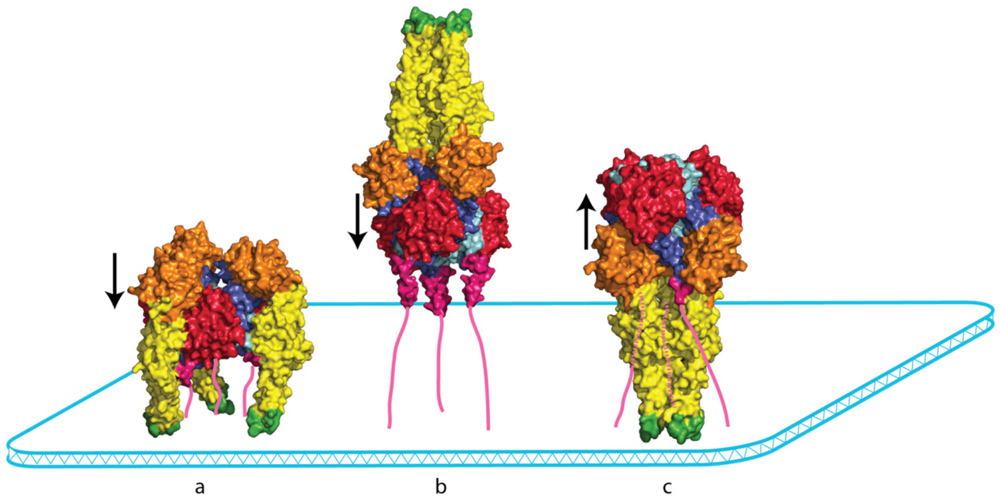
5.1. Crystallographic Structure of the Pre- and Post-Fusion States of the VSV G Ectodomain

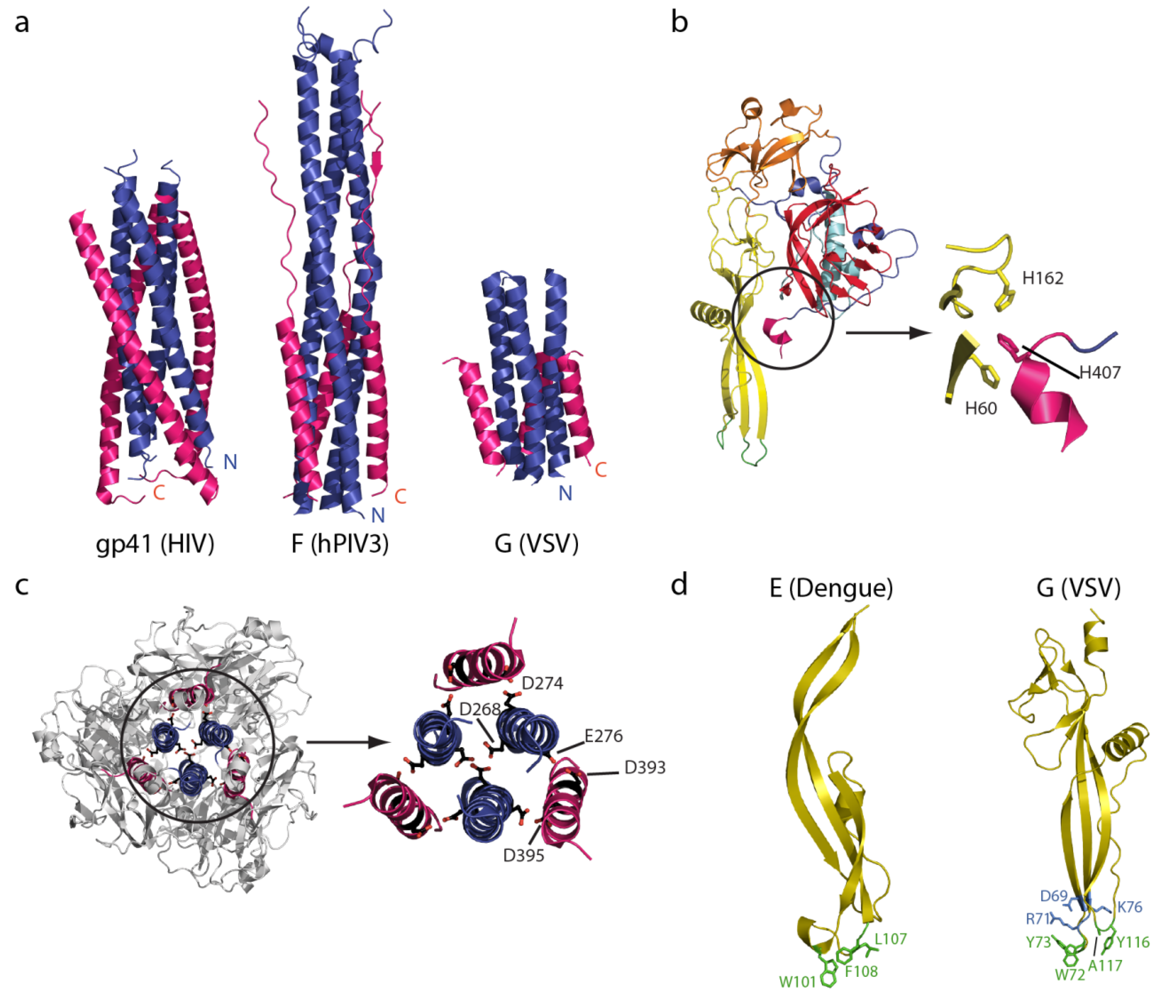
5.2. The Structural Transition
5.3. Interaction between G and Membranes

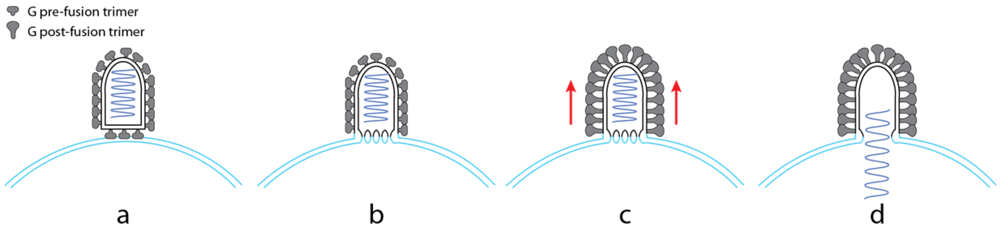
5.4. Cooperativity between Glycoproteins during the Fusion Process

5.5. Intermediates during the Conformational Change

6. Cellular Biology of Rhabdovirus Entry into the Host Cell
6.1. Cellular Aspects of VSV Entry

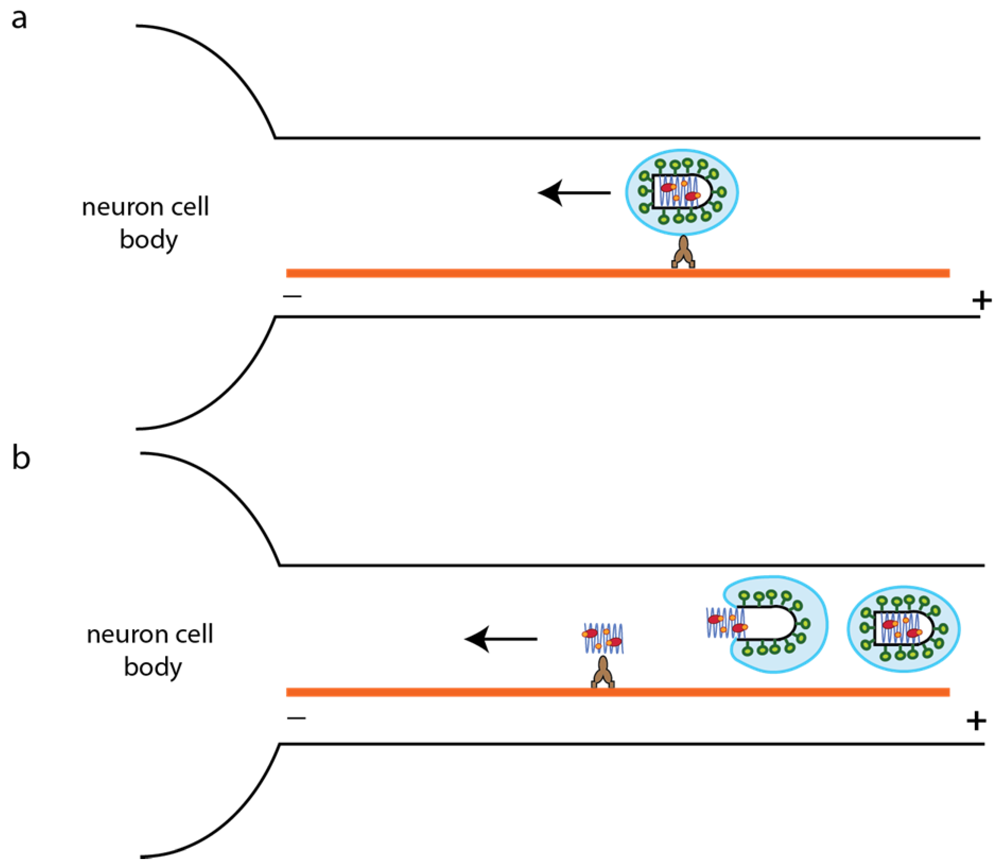
6.2. RABV Entry into Neurons

6.3. Early Post Entry Events
7. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Rose, J.K.; Whitt, M.A. Rhabdoviridae: The viruses and their replication. In Fields Virology, 4th ed; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2001; pp. 1221–1224. [Google Scholar]
- Fort, P.; Albertini, A.; Van-Hua, A.; Berthomieu, A.; Roche, S.; Delsuc, F.; Pasteur, N.; Capy, P.; Gaudin, Y.; Weill, M. Fossil rhabdoviral sequences integrated into arthropod genomes: Ontogeny, evolution, and potential functionality. Mol. Biol. Evol. 2011. [Google Scholar]
- Mebatsion, T.; Weiland, F.; Conzelmann, K.K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 1999, 73, 242–250. [Google Scholar]
- Doms, R.W.; Keller, D.S.; Helenius, A.; Balch, W.E. Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J. Cell Biol. 1987, 105, 1957–1969. [Google Scholar]
- Gaudin, Y.; Ruigrok, R.W.; Tuffereau, C.; Knossow, M.; Flamand, A. Rabies virus glycoprotein is a trimer. Virology 1992, 187, 627–632. [Google Scholar]
- Whitt, M.A.; Buonocore, L.; Prehaud, C.; Rose, J.K. Membrane fusion activity, oligomerization, and assembly of the rabies virus glycoprotein. Virology 1991, 185, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Elliott, M.M.; Keller, D.S. ATP-coupled transport of vesicular stomatitis virus G protein between the endoplasmic reticulum and the Golgi. J. Biol. Chem. 1986, 261, 14681–14689. [Google Scholar]
- Kreis, T.E.; Lodish, H.F. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell 1986, 46, 929–937. [Google Scholar]
- Lyles, D.S.; Varela, V.A.; Parce, J.W. Dynamic nature of the quaternary structure of the vesicular stomatitis virus envelope glycoprotein. Biochemistry 1990, 29, 2442–2449. [Google Scholar]
- Wilcox, M.D.; McKenzie, M.O.; Parce, J.W.; Lyles, D.S. Subunit interactions of vesicular stomatitis virus envelope glycoprotein influenced by detergent micelles and lipid bilayers. Biochemistry 1992, 31, 10458–10464. [Google Scholar]
- Zagouras, P.; Ruusala, A.; Rose, J.K. Dissociation and reassociation of oligomeric viral glycoprotein subunits in the endoplasmic reticulum. J. Virol. 1991, 65, 1976–1984. [Google Scholar]
- Gaudin, Y. Folding of rabies virus glycoprotein: Epitope acquisition and interaction with endoplasmic reticulum chaperones. J. Virol. 1997, 71, 3742–3750. [Google Scholar]
- Hammond, C.; Helenius, A. Folding of VSV G protein: Sequential interaction with BiP and calnexin. Science 1994, 266, 456–458. [Google Scholar]
- Mathieu, M.E.; Grigera, P.R.; Helenius, A.; Wagner, R.R. Folding, unfolding, and refolding of the vesicular stomatitis virus glycoprotein. Biochemistry 1996, 35, 4084–4093. [Google Scholar] [PubMed]
- Flamand, A.; Raux, H.; Gaudin, Y.; Ruigrok, R.W. Mechanisms of rabies virus neutralization. Virology 1993, 194, 302–313. [Google Scholar]
- Lefrancois, L.; Lyles, D.S. Antigenic determinants of vesicular stomatitis virus: analysis with antigenic variants. J. Immunol. 1983, 130, 394–398. [Google Scholar]
- Prehaud, C.; Coulon, P.; LaFay, F.; Thiers, C.; Flamand, A. Antigenic site II of the rabies virus glycoprotein: structure and role in viral virulence. J. Virol. 1988, 62, 1–7. [Google Scholar]
- Seif, I.; Coulon, P.; Rollin, P.E.; Flamand, A. Rabies virulence: effect on pathogenicity and sequence characterization of rabies virus mutations affecting antigenic site III of the glycoprotein. J. Virol. 1985, 53, 926–934. [Google Scholar]
- Vandepol, S.B.; Lefrancois, L.; Holland, J.J. Sequences of the major antibody binding epitopes of the Indiana serotype of vesicular stomatitis virus. Virology 1986, 148, 312–325. [Google Scholar]
- Coulon, P.; Ternaux, J.P.; Flamand, A.; Tuffereau, C. An avirulent mutant of rabies virus is unable to infect motoneurons in vivo and in vitro. J. Virol. 1998, 72, 273–278. [Google Scholar] [PubMed]
- Tuffereau, C.; Leblois, H.; Benejean, J.; Coulon, P.; Lafay, F.; Flamand, A. Arginine or lysine in position 333 of ERA and CVS glycoprotein is necessary for rabies virulence in adult mice. Virology 1989, 172, 206–212. [Google Scholar]
- Benmansour, A.; Leblois, H.; Coulon, P.; Tuffereau, C.; Gaudin, Y.; Flamand, A.; Lafay, F. Antigenicity of rabies virus glycoprotein. J. Virol. 1991, 65, 4198–4203. [Google Scholar]
- Lafay, F.; Benmansour, A.; Chebli, K.; Flamand, A. Immunodominant epitopes defined by a yeast-expressed library of random fragments of the rabies virus glycoprotein map outside major antigenic sites. J. Gen. Virol. 1996, 77, 339–346. [Google Scholar]
- Raux, H.; Coulon, P.; Lafay, F.; Flamand, A. Monoclonal antibodies which recognize the acidic configuration of the rabies glycoprotein at the surface of the virion can be neutralizing. Virology 1995, 210, 400–408. [Google Scholar]
- Dietzschold, B.; Gore, M.; Marchadier, D.; Niu, H.S.; Bunschoten, H.M.; Otvos, L., Jr.; Wunner, W.H.; Ertl, H.C.; Osterhaus, A.D.; Koprowski, H. Structural and immunological characterization of a linear virus-neutralizing epitope of the rabies virus glycoprotein and its possible use in a synthetic vaccine. J. Virol. 1990, 64, 3804–3809. [Google Scholar]
- Lafon, M.; Wiktor, T.J.; Macfarlan, R.I. Antigenic sites on the CVS rabies virus glycoprotein: analysis with monoclonal antibodies. J. Gen. Virol. 1983, 64, 843–851. [Google Scholar]
- Schlegel, R.; Tralka, T.S.; Willingham, M.C.; Pastan, I. Inhibition of VSV binding and infectivity by phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell 1983, 32, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Coil, D.A.; Miller, A.D. Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J. Virol. 2004, 78, 10920–10926. [Google Scholar]
- Sinibaldi, L.; Goldoni, P.; Seganti, L.; Superti, F.; Tsiang, H.; Orsi, N. Gangliosides in early interactions between vesicular stomatitis virus and CER cells. Microbiologica 1985, 8, 355–365. [Google Scholar]
- Bloor, S.; Maelfait, J.; Krumbach, R.; Beyaert, R.; Randow, F. Endoplasmic reticulum chaperone gp96 is essential for infection with vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6970–6975. [Google Scholar]
- Kissling, R.E. Growth of rabies virus in non-nervous tissue culture. Proc. Soc. Exp. Biol. Med. 1958, 98, 223–225. [Google Scholar]
- Wiktor, T.J.; Fernandes, M.V.; Koprowski, H. Cultivation Of rabies virus in human diploid cell strain Wi-38. J. Immunol. 1964, 93, 353–366. [Google Scholar]
- Schneider, L.G.; Horzinek, M.; Matheka, H.D. Purification of rabies virus from tissue culture. Arch. Gesamte Virusforsch. 1971, 34, 351–359. [Google Scholar]
- Seganti, L.; Superti, F.; Bianchi, S.; Orsi, N.; Divizia, M.; Pana, A. Susceptibility of mammalian, avian, fish, and mosquito cell lines to rabies virus infection. Acta Virol. 1990, 34, 155–163. [Google Scholar] [PubMed]
- Lafon, M. Rabies virus receptors. J. Neurovirol. 2005, 11, 82–87. [Google Scholar]
- Superti, F.; Seganti, L.; Tsiang, H.; Orsi, N. Role of phospholipids in rhabdovirus attachment to CER cells. Brief report. Arch. Virol. 1984, 81, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Conti, C.; Superti, F.; Tsiang, H. Membrane carbohydrate requirement for rabies virus binding to chicken embryo related cells. Intervirology 1986, 26, 164–168. [Google Scholar]
- Superti, F.; Hauttecoeur, B.; Morelec, M.J.; Goldoni, P.; Bizzini, B.; Tsiang, H. Involvement of gangliosides in rabies virus infection. J. Gen. Virol. 1986, 67, 47–56. [Google Scholar]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [PubMed]
- Lentz, T.L.; Wilson, P.T.; Hawrot, E.; Speicher, D.W. Amino acid sequence similarity between rabies virus glycoprotein and snake venom curaremimetic neurotoxins. Science 1984, 226, 847–848. [Google Scholar]
- Lentz, T.L.; Benson, R.J.; Klimowicz, D.; Wilson, P.T.; Hawrot, E. Binding of rabies virus to purified Torpedo acetylcholine receptor. Brain Res. 1986, 387, 211–219. [Google Scholar]
- Gastka, M.; Horvath, J.; Lentz, T.L. Rabies virus binding to the nicotinic acetylcholine receptor alpha subunit demonstrated by virus overlay protein binding assay. J. Gen. Virol. 1996, 77, 2437–2440. [Google Scholar]
- McGehee, D.S.; Role, L.W. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu. Rev. Physiol. 1995, 57, 521–546. [Google Scholar]
- Burrage, T.G.; Tignor, G.H.; Smith, A.L. Rabies virus binding at neuromuscular junctions. Virus Res. 1985, 2, 273–289. [Google Scholar]
- Thoulouze, M.I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The neural cell adhesion molecule is a receptor for rabies virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar]
- Tuffereau, C.; Benejean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar]
- Langevin, C.; Tuffereau, C. Mutations conferring resistance to neutralization by a soluble form of the neurotrophin receptor (p75NTR) map outside of the known antigenic sites of the rabies virus glycoprotein. J. Virol. 2002, 76, 10756–10765. [Google Scholar]
- Tuffereau, C.; Desmezieres, E.; Benejean, J.; Jallet, C.; Flamand, A.; Tordo, N.; Perrin, P. Interaction of lyssaviruses with the low-affinity nerve-growth factor receptor p75NTR. J. Gen. Virol. 2001, 82, 2861–2867. [Google Scholar]
- Tuffereau, C.; Schmidt, K.; Langevin, C.; Lafay, F.; Dechant, G.; Koltzenburg, M. The rabies virus glycoprotein receptor p75NTR is not essential for rabies virus infection. J. Virol. 2007, 81, 13622–13630. [Google Scholar]
- Bearzotti, M.; Delmas, B.; Lamoureux, A.; Loustau, A.M.; Chilmonczyk, S.; Bremont, M. Fish rhabdovirus cell entry is mediated by fibronectin. J. Virol. 1999, 73, 7703–7709. [Google Scholar]
- Roche, S.; Albertini, A.A.; Lepault, J.; Bressanelli, S.; Gaudin, Y. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell Mol. Life Sci. 2008, 65, 1716–1728. [Google Scholar]
- Florkiewicz, R.Z.; Rose, J.K. A cell line expressing vesicular stomatitis virus glycoprotein fuses at low pH. Science 1984, 225, 721–723. [Google Scholar]
- Riedel, H.; Kondor-Koch, C.; Garoff, H. Cell surface expression of fusogenic vesicular stomatitis virus G protein from cloned cDNA. EMBO J. 1984, 3, 1477–1483. [Google Scholar]
- Clague, M.J.; Schoch, C.; Zech, L.; Blumenthal, R. Gating kinetics of pH-activated membrane fusion of vesicular stomatitis virus with cells: Stopped-flow measurements by dequenching of octadecylrhodamine fluorescence. Biochemistry 1990, 29, 1303–1308. [Google Scholar]
- Gaudin, Y.; de Kinkelin, P.; Benmansour, A. Mutations in the glycoprotein of viral haemorrhagic septicaemia virus that affect virulence for fish and the pH threshold for membrane fusion. J. Gen. Virol. 1999, 80, 1221–1229. [Google Scholar]
- Roche, S.; Gaudin, Y. Evidence that rabies virus forms different kinds of fusion machines with different pH thresholds for fusion. J. Virol. 2004, 78, 8746–8752. [Google Scholar]
- Gaudin, Y. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 2000, 34, 379–408. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, Y.; Ruigrok, R.W.; Knossow, M.; Flamand, A. Low-pH conformational changes of rabies virus glycoprotein and their role in membrane fusion. J. Virol. 1993, 67, 1365–1372. [Google Scholar]
- Gaudin, Y.; Tuffereau, C.; Segretain, D.; Knossow, M.; Flamand, A. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J. Virol. 1991, 65, 4853–4859. [Google Scholar]
- Durrer, P.; Gaudin, Y.; Ruigrok, R.W.; Graf, R.; Brunner, J. Photolabeling identifies a putative fusion domain in the envelope glycoprotein of rabies and vesicular stomatitis viruses. J. Biol. Chem. 1995, 270, 17575–17581. [Google Scholar]
- Roche, S.; Gaudin, Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 2002, 297, 128–135. [Google Scholar]
- Gaudin, Y.; Tuffereau, C.; Durrer, P.; Flamand, A.; Ruigrok, R.W. Biological function of the low-pH, fusion-inactive conformation of rabies virus glycoprotein (G): G is transported in a fusion-inactive state-like conformation. J. Virol. 1995, 69, 5528–5534. [Google Scholar]
- Albertini, A.; Bressanelli, S.; Lepault, J.; Gaudin, Y. Structure and working of viral fusion machinery. Curr. Top. Membr. 2011, 68, 49–80. [Google Scholar]
- Carr, C.M.; Chaudhry, C.; Kim, P.S. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 14306–14313. [Google Scholar]
- Kuzmin, P.I.; Zimmerberg, J.; Chizmadzhev, Y.A.; Cohen, F.S. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7235–7240. [Google Scholar]
- Lee, J.; Lentz, B.R. Secretory and viral fusion may share mechanistic events with fusion between curved lipid bilayers. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 9274–9279. [Google Scholar]
- Herrmann, A.; Clague, M.J.; Puri, A.; Morris, S.J.; Blumenthal, R.; Grimaldi, S. Effect of erythrocyte transbilayer phospholipid distribution on fusion with vesicular stomatitis virus. Biochemistry 1990, 29, 4054–4058. [Google Scholar]
- Yamada, S.; Ohnishi, S. Vesicular stomatitis virus binds and fuses with phospholipid domain in target cell membranes. Biochemistry 1986, 25, 3703–3708. [Google Scholar]
- Roth, S.L.; Whittaker, G.R. Promotion of vesicular stomatitis virus fusion by the endosome-specific phospholipid bis(monoacylglycero)phosphate (BMP). FEBS Lett. 2011, 585, 865–869. [Google Scholar]
- Chernomordik, L.V.; Frolov, V.A.; Leikina, E.; Bronk, P.; Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998, 140, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Zaitseva, E.; Mittal, A.; Griffin, D.E.; Chernomordik, L.V. Class II fusion protein of alphaviruses drives membrane fusion through the same pathway as class I proteins. J. Cell Biol. 2005, 169, 167–177. [Google Scholar]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein g. Science 2007, 315, 843–848. [Google Scholar]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2880–2885. [Google Scholar]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar]
- Carneiro, F.A.; Stauffer, F.; Lima, C.S.; Juliano, M.A.; Juliano, L.; Da Poian, A.T. Membrane fusion induced by vesicular stomatitis virus depends on histidine protonation. J. Biol. Chem. 2003, 278, 13789–13794. [Google Scholar]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar]
- Fredericksen, B.L.; Whitt, M.A. Vesicular stomatitis virus glycoprotein mutations that affect membrane fusion activity and abolish virus infectivity. J. Virol. 1995, 69, 1435–1443. [Google Scholar]
- Rocha, A.; Ruiz, S.; Tafalla, C.; Coll, J.M. Conformation- and fusion-defective mutations in the hypothetical phospholipid-binding and fusion peptides of viral hemorrhagic septicemia salmonid rhabdovirus protein G. J. Virol. 2004, 78, 9115–9122. [Google Scholar]
- Stanifer, M.L.; Cureton, D.K.; Whelan, S.P. A recombinant vesicular stomatitis virus bearing a lethal mutation in the glycoprotein gene uncovers a second site suppressor that restores fusion. J. Virol. 2011, 85, 8105–8115. [Google Scholar]
- Sun, X.; Belouzard, S.; Whittaker, G.R. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J. Biol. Chem. 2008, 283, 6418–6427. [Google Scholar]
- Wimley, W.C.; White, S.H. Partitioning of tryptophan side-chain analogs between water and cyclohexane. Biochemistry 1992, 31, 12813–12818. [Google Scholar]
- Efrat, A.; Chernomordik, L.V.; Kozlov, M.M. Point-like protrusion as a prestalk intermediate in membrane fusion pathway. Biophys. J. 2007, 92, L61–63. [Google Scholar]
- Kemble, G.W.; Danieli, T.; White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994, 76, 383–391. [Google Scholar]
- Odell, D.; Wanas, E.; Yan, J.; Ghosh, H.P. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 1997, 71, 7996–8000. [Google Scholar]
- Cleverley, D.Z.; Lenard, J. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 3425–3430. [Google Scholar]
- Jeetendra, E.; Ghosh, K.; Odell, D.; Li, J.; Ghosh, H.P.; Whitt, M.A. The membrane-proximal region of vesicular stomatitis virus glycoprotein G ectodomain is critical for fusion and virus infectivity. J. Virol. 2003, 77, 12807–12818. [Google Scholar]
- Maillard, A.; Domanski, M.; Brunet, P.; Chaffotte, A.; Guittet, E.; Gaudin, Y. Spectroscopic characterization of two peptides derived from the stem of rabies virus glycoprotein. Virus Res. 2003, 93, 151–158. [Google Scholar]
- Libersou, S.; Albertini, A.A.; Ouldali, M.; Maury, V.; Maheu, C.; Raux, H.; de Haas, F.; Roche, S.; Gaudin, Y.; Lepault, J. Distinct structural rearrangements of the VSV glycoprotein drive membrane fusion. J. Cell Biol. 2010, 191, 199–210. [Google Scholar]
- Gaudin, Y.; Raux, H.; Flamand, A.; Ruigrok, R.W. Identification of amino acids controlling the low-pH-induced conformational change of rabies virus glycoprotein. J. Virol. 1996, 70, 7371–7378. [Google Scholar]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar]
- Weissenhorn, W.; Hinz, A.; Gaudin, Y. Virus membrane fusion. FEBS Lett. 2007, 581, 2150–2155. [Google Scholar]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar]
- Johannsdottir, H.K.; Mancini, R.; Kartenbeck, J.; Amato, L.; Helenius, A. Host cell factors and functions involved in vesicular stomatitis virus entry. J. Virol. 2009, 83, 440–453. [Google Scholar]
- Superti, F.; Seganti, L.; Ruggeri, F.M.; Tinari, A.; Donelli, G.; Orsi, N. Entry pathway of vesicular stomatitis virus into different host cells. J. Gen. Virol. 1987, 68, 387–399. [Google Scholar]
- Sun, X.; Yau, V.K.; Briggs, B.J.; Whittaker, G.R. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 2005, 338, 53–60. [Google Scholar]
- Cureton, D.K.; Massol, R.H.; Saffarian, S.; Kirchhausen, T.L.; Whelan, S.P. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009, 5, e1000394. [Google Scholar]
- Cureton, D.K.; Massol, R.H.; Whelan, S.P.; Kirchhausen, T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010, 6, e1001127. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 2003, 4, 333–343. [Google Scholar]
- Mire, C.E.; White, J.M.; Whitt, M.A. A spatio-temporal analysis of matrix protein and nucleocapsid trafficking during vesicular stomatitis virus uncoating. PLoS Pathog. 2010, 6, e1000994. [Google Scholar]
- Le Blanc, I.; Luyet, P.P.; Pons, V.; Ferguson, C.; Emans, N.; Petiot, A.; Mayran, N.; Demaurex, N.; Faure, J.; Sadoul, R.; et al. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 2005, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Raux, H.; Flamand, A.; Blondel, D. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 2000, 74, 10212–10216. [Google Scholar]
- Jacob, Y.; Badrane, H.; Ceccaldi, P.E.; Tordo, N. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 2000, 74, 10217–10222. [Google Scholar]
- Poisson, N.; Real, E.; Gaudin, Y.; Vaney, M.C.; King, S.; Jacob, Y.; Tordo, N.; Blondel, D. Molecular basis for the interaction between rabies virus phosphoprotein P and the dynein light chain LC8: dissociation of dynein-binding properties and transcriptional functionality of P. J. Gen. Virol. 2001, 82, 2691–2696. [Google Scholar]
- Mebatsion, T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J. Virol. 2001, 75, 11496–11502. [Google Scholar]
- Tan, G.S.; Preuss, M.A.; Williams, J.C.; Schnell, M.J. The dynein light chain 8 binding motif of rabies virus phosphoprotein promotes efficient viral transcription. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 7229–7234. [Google Scholar]
- Mazarakis, N.D.; Azzouz, M.; Rohll, J.B.; Ellard, F.M.; Wilkes, F.J.; Olsen, A.L.; Carter, E.E.; Barber, R.D.; Baban, D.F.; Kingsman, S.M.; et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001, 10, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Mentis, G.Z.; Gravell, M.; Hamilton, R.; Shneider, N.A.; O'Donovan, M.J.; Schubert, M. Transduction of motor neurons and muscle fibers by intramuscular injection of HIV-1-based vectors pseudotyped with select rabies virus glycoproteins. J. Neurosci. Meth. 2006, 157, 208–217. [Google Scholar]
- Klingen, Y.; Conzelmann, K.K.; Finke, S. Double-labeled rabies virus: Live tracking of enveloped virus transport. J. Virol. 2008, 82, 237–245. [Google Scholar]
- Mire, C.E.; Dube, D.; Delos, S.E.; White, J.M.; Whitt, M.A. Glycoprotein-dependent acidification of vesicular stomatitis virus enhances release of matrix protein. J. Virol. 2009, 83, 12139–12150. [Google Scholar]
- Kasermann, F.; Kempf, C. Low pH-induced pore formation by spike proteins of enveloped viruses. J. Gen. Virol. 1996, 77, 3025–3032. [Google Scholar]
- Menager, P.; Roux, P.; Megret, F.; Bourgeois, J.P.; Le Sourd, A.M.; Danckaert, A.; Lafage, M.; Prehaud, C.; Lafon, M. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog. 2009, 5, e1000315. [Google Scholar]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar]
- Das, S.C.; Nayak, D.; Zhou, Y.; Pattnaik, A.K. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 2006, 80, 6368–6377. [Google Scholar]
- Heinrich, B.S.; Cureton, D.K.; Rahmeh, A.A.; Whelan, S.P. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 2010, 6, e1000958. [Google Scholar]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 2011. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Albertini, A.A.V.; Baquero, E.; Ferlin, A.; Gaudin, Y. Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses 2012, 4, 117-139. https://doi.org/10.3390/v4010117
Albertini AAV, Baquero E, Ferlin A, Gaudin Y. Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses. 2012; 4(1):117-139. https://doi.org/10.3390/v4010117
Chicago/Turabian StyleAlbertini, Aurélie A. V., Eduard Baquero, Anna Ferlin, and Yves Gaudin. 2012. "Molecular and Cellular Aspects of Rhabdovirus Entry" Viruses 4, no. 1: 117-139. https://doi.org/10.3390/v4010117
APA StyleAlbertini, A. A. V., Baquero, E., Ferlin, A., & Gaudin, Y. (2012). Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses, 4(1), 117-139. https://doi.org/10.3390/v4010117