Filovirus Entry: A Novelty in the Viral Fusion World

{kind=link}
{kind=link}
Abstract
:1. Introduction
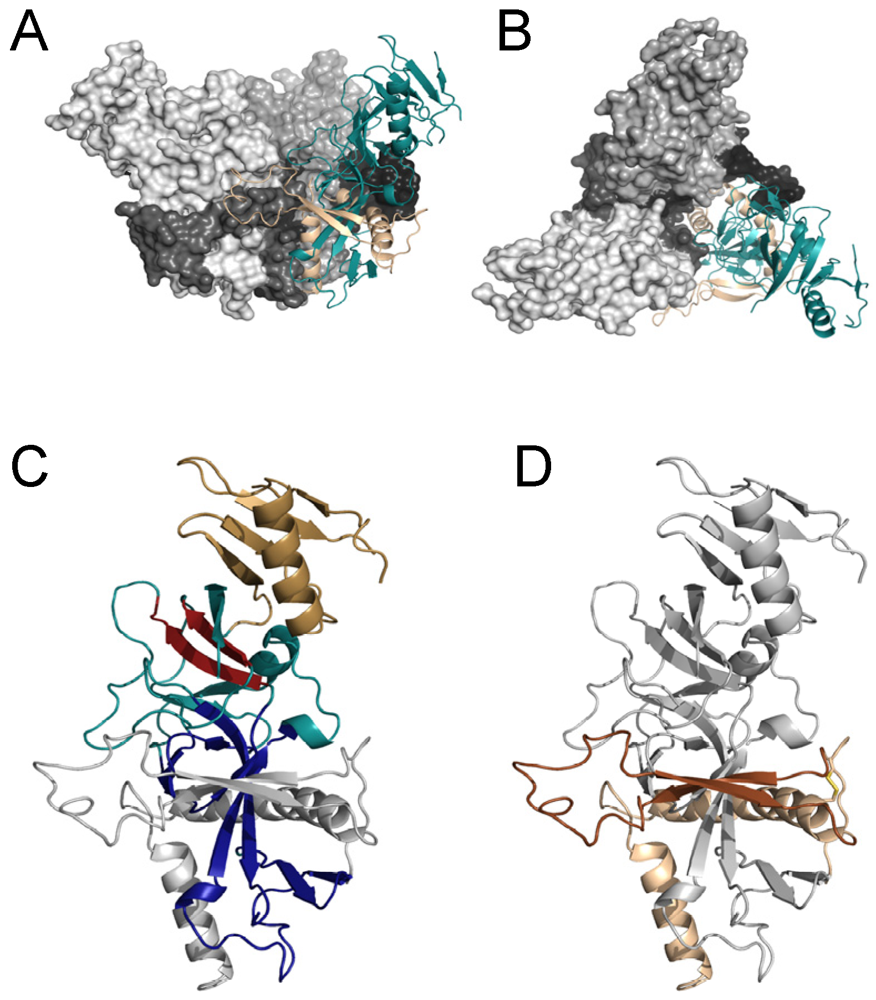
3. Structure of Filovirus GPs
4. GP1

5. GP2
6. The Role of Cell Surface Proteins in Filovirus GP-Dependent Entry
6.1. C-Type Lectins
6.2. β1 Integrins
6.3. Tyro3 (TAM) Family Tyrosine Kinase Receptors
6.4. TIM-1
8. Proteolytic Processing of EBOV GP1,2 into a Fusion-Active Form
9. NPC1
10. Characterized Filovirus Fusion Events
11. Potential Therapeutics against Filovirus Entry

Acknowledgments
Conflict of Interest
References and Notes
- Falzarano, D.; Krokhin, O.; Wahl-Jensen, V.; Seebach, J.; Wolf, K.; Schnittler, H.J.; Feldmann, H. Structure-function analysis of the soluble glycoprotein, sgp, of ebola virus. Chembiochem 2006, 7, 1605–1611. [Google Scholar] [CrossRef]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.J.; Feldmann, H. A new ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar]
- Neumann, G.; Feldmann, H.; Watanabe, S.; Lukashevich, I.; Kawaoka, Y. Reverse genetics demonstrates that proteolytic processing of the ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 2002, 76, 406–410. [Google Scholar]
- Sanchez, A.; Yang, Z.Y.; Xu, L.; Nabel, G.J.; Crews, T.; Peters, C.J. Biochemical analysis of the secreted and virion glycoproteins of ebola virus. J. Virol. 1998, 72, 6442–6447. [Google Scholar]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein gp2 from ebola virus at 1.9-a resolution. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 2662–2667. [Google Scholar]
- Weissenhorn, W.; Carfi, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the ebola virus membrane fusion subunit, gp2, from the envelope glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Radoshitzky, S.R.; Guth, A.C.; Warfield, K.L.; Li, W.; Vincent, M.J.; Towner, J.S.; Nichol, S.T.; Bavari, S.; Choe, H.; et al. Conserved receptor-binding domains of lake victoria marburgvirus and zaire ebolavirus bind a common receptor. J. Biol. Chem. 2006, 281, 15951–15958. [Google Scholar]
- Manicassamy, B.; Wang, J.; Rumschlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.; Rong, L. Characterization of marburg virus glycoprotein in viral entry. Virology 2007, 358, 79–88. [Google Scholar]
- Brindley, M.A.; Hughes, L.; Ruiz, A.; McCray, P.B., Jr.; Sanchez, A.; Sanders, D.A.; Maury, W. Ebola virus glycoprotein 1: Identification of residues important for binding and postbinding events. J. Virol. 2007, 81, 7702–7709. [Google Scholar]
- Dube, D.; Brecher, M.B.; Delos, S.E.; Rose, S.C.; Park, E.W.; Schornberg, K.L.; Kuhn, J.H.; White, J.M. The primed ebolavirus glycoprotein (19-kilodalton gp1,2): Sequence and residues critical for host cell binding. J. Virol. 2009, 83, 2883–2891. [Google Scholar]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent modifications of the ebola virus glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar]
- Sullivan, N.J.; Peterson, M.; Yang, Z.Y.; Kong, W.P.; Duckers, H.; Nabel, E.; Nabel, G.J. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 2005, 79, 547–553. [Google Scholar]
- Jaskierny, A.J.; Panahi, A.; Feig, M. Effect of flanking residues on the conformational sampling of the internal fusion peptide from ebola virus. Proteins 2011, 79, 1109–1117. [Google Scholar]
- Watanabe, S.; Takada, A.; Watanabe, T.; Ito, H.; Kida, H.; Kawaoka, Y. Functional importance of the coiled-coil of the ebola virus glycoprotein. J. Virol. 2000, 74, 10194–10201. [Google Scholar]
- Usami, K.; Matsuno, K.; Igarashi, M.; Denda-Nagai, K.; Takada, A.; Irimura, T. Involvement of viral envelope gp2 in ebola virus entry into cells expressing the macrophage galactose-type c-type lectin. Biochem. Biophys. Res. Commun. 2011, 407, 74–78. [Google Scholar]
- Han, Z.; Licata, J.M.; Paragas, J.; Harty, R.N. Permeabilization of the plasma membrane by ebola virus gp2. Virus Genes 2007, 34, 273–281. [Google Scholar]
- Adam, B.; Lins, L.; Stroobant, V.; Thomas, A.; Brasseur, R. Distribution of hydrophobic residues is crucial for the fusogenic properties of the ebola virus gp2 fusion peptide. J. Virol. 2004, 78, 2131–2136. [Google Scholar]
- Ito, H.; Watanabe, S.; Sanchez, A.; Whitt, M.A.; Kawaoka, Y. Mutational analysis of the putative fusion domain of ebola virus glycoprotein. J. Virol. 1999, 73, 8907–8912. [Google Scholar]
- Ruiz-Arguello, M.B.; Goni, F.M.; Pereira, F.B.; Nieva, J.L. Phosphatidylinositol-dependent membrane fusion induced by a putative fusogenic sequence of ebola virus. J. Virol. 1998, 72, 1775–1781. [Google Scholar]
- Plemper, R.K. Cell entry of enveloped viruses. Curr. Opin. Virol. 2011, 1, 92–100. [Google Scholar]
- Schroth-Diez, B.; Ludwig, K.; Baljinnyam, B.; Kozerski, C.; Huang, Q.; Herrmann, A. The role of the transmembrane and of the intraviral domain of glycoproteins in membrane fusion of enveloped viruses. Biosci. Rep. 2000, 20, 571–595. [Google Scholar]
- Ito, H.; Watanabe, S.; Takada, A.; Kawaoka, Y. Ebola virus glycoprotein: Proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 2001, 75, 1576–1580. [Google Scholar] [CrossRef]
- Feldmann, H.; Volchkov, V.E.; Volchkova, V.A.; Klenk, H.D. The glycoproteins of marburg and ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 1999, 15, 159–169. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E. Ebola virus: New insights into disease aetiopathology and possible therapeutic interventions. Expert Rev. Mol. Med. 2004, 6, 1–24. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of ebola hemorrhagic fever in cynomolgus macaques: Evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar]
- Wool-Lewis, R.J.; Bates, P. Characterization of ebola virus entry by using pseudotyped viruses: Identification of receptor-deficient cell lines. J. Virol. 1998, 72, 3155–3160. [Google Scholar]
- Yang, Z.; Delgado, R.; Xu, L.; Todd, R.F.; Nabel, E.G.; Sanchez, A.; Nabel, G.J. Distinct cellular interactions of secreted and transmembrane ebola virus glycoproteins. Science 1998, 279, 1034–1037. [Google Scholar]
- Breman, J.G.; Johnson, K.M.; van der Groen, G.; Robbins, C.B.; Szczeniowski, M.V.; Ruti, K.; Webb, P.A.; Meier, F.; Heymann, D.L. A search for ebola virus in animals in the democratic republic of the Congo and Cameroon: Ecologic, virologic, and serologic surveys, 1979–1980. Ebola virus study teams. J. Infect. Dis. 1999, 179, S139–S147. [Google Scholar]
- Chan, S.Y.; Speck, R.F.; Ma, M.C.; Goldsmith, M.A. Distinct mechanisms of entry by envelope glycoproteins of marburg and ebola (zaire) viruses. J. Virol. 2000, 74, 4933–4937. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by ebola and marburg viruses. Lab. Invest. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jahrling, P.B.; Hanes, M.A.; Zack, P.M. Association of ebola-related reston virus particles and antigen with tissue lesions of monkeys imported to the united states. J. Comp. Pathol. 1992, 106, 137–152. [Google Scholar]
- Simmons, G.; Rennekamp, A.J.; Chai, N.; Vandenberghe, L.H.; Riley, J.L.; Bates, P. Folate receptor alpha and caveolae are not required for ebola virus glycoprotein-mediated viral infection. J. Virol. 2003, 77, 13433–13438. [Google Scholar]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. Dc-sign and dc-signr bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar]
- Albritton, L.M.; Tseng, L.; Scadden, D.; Cunningham, J.M. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell 1989, 57, 659–666. [Google Scholar]
- O'Hara, B.; Johann, S.V.; Klinger, H.P.; Blair, D.G.; Rubinson, H.; Dunn, K.J.; Sass, P.; Vitek, S.M.; Robins, T. Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ. 1990, 1, 119–127. [Google Scholar]
- Bates, P.; Young, J.A.; Varmus, H.E. A receptor for subgroup a rous sarcoma virus is related to the low density lipoprotein receptor. Cell 1993, 74, 1043–1051. [Google Scholar]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of ebola and marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar]
- Chan, S.Y.; Empig, C.J.; Welte, F.J.; Speck, R.F.; Schmaljohn, A.; Kreisberg, J.F.; Goldsmith, M.A. Folate receptor-alpha is a cofactor for cellular entry by marburg and ebola viruses. Cell 2001, 106, 117–126. [Google Scholar]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (tim-1) is a receptor for zaire ebolavirus and lake victoria marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 8426–8431. [Google Scholar]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muniz, O.; Corbi, A.L.; Delgado, R. C-type lectins dc-sign and l-sign mediate cellular entry by ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar]
- Becker, S.; Spiess, M.; Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for marburg virus. J. Gen. Virol. 1995, 76, 393–399. [Google Scholar]
- Gramberg, T.; Hofmann, H.; Moller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. Lsectin interacts with filovirus glycoproteins and the spike protein of sars coronavirus. Virology 2005, 340, 224–236. [Google Scholar]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human macrophage c-type lectin specific for galactose and n-acetylgalactosamine promotes filovirus entry. J. Virol. 2004, 78, 2943–2947. [Google Scholar]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. Dc-sign and dc-signr interact with the glycoprotein of marburg virus and the s protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar]
- Marzi, A.; Wegele, A.; Pohlmann, S. Modulation of virion incorporation of ebolavirus glycoprotein: Effects on attachment, cellular entry and neutralization. Virology 2006, 352, 345–356. [Google Scholar]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar]
- Kaletsky, R.L.; Simmons, G.; Bates, P. Proteolysis of the ebola virus glycoproteins enhances virus binding and infectivity. J. Virol. 2007, 81, 13378–13384. [Google Scholar]
- Matsuno, K.; Nakayama, E.; Noyori, O.; Marzi, A.; Ebihara, H.; Irimura, T.; Feldmann, H.; Takada, A. C-type lectins do not act as functional receptors for filovirus entry into cells. Biochem. Biophys. Res. Commun. 2010, 403, 144–148. [Google Scholar]
- Tsegaye, T.S.; Pohlmann, S. The multiple facets of HIV attachment to dendritic cell lectins. Cell Microbiol. 2010, 12, 1553–1561. [Google Scholar]
- Simmons, G.; Wool-Lewis, R.J.; Baribaud, F.; Netter, R.C.; Bates, P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002, 76, 2518–2528. [Google Scholar]
- Takada, A.; Watanabe, S.; Ito, H.; Okazaki, K.; Kida, H.; Kawaoka, Y. Downregulation of beta1 integrins by ebola virus glycoprotein: Implication for virus entry. Virology 2000, 278, 20–26. [Google Scholar]
- Schornberg, K.L.; Shoemaker, C.J.; Dube, D.; Abshire, M.Y.; Delos, S.E.; Bouton, A.H.; White, J.M. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8003–8008. [Google Scholar]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Tam receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of axl-mediated ebola virus infection. J. Infect. Dis. 2007, 196, S259–S263. [Google Scholar]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B., Jr.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor axl enhances entry of zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The tyro3 receptor kinase axl enhances macropinocytosis of zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6. [Google Scholar]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6. [Google Scholar]
- Weller, M.L.; Amornphimoltham, P.; Schmidt, M.; Wilson, P.A.; Gutkind, J.S.; Chiorini, J.A. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 2010, 16, 662–664. [Google Scholar]
- Yonezawa, A.; Cavrois, M.; Greene, W.C. Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: Involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J. Virol. 2005, 79, 918–926. [Google Scholar]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid raft microdomains: A gateway for compartmentalized trafficking of ebola and marburg viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar]
- Empig, C.J.; Goldsmith, M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002, 76, 5266–5270. [Google Scholar]
- Bhattacharyya, S.; Hope, T.J.; Young, J.A. Differential requirements for clathrin endocytic pathway components in cellular entry by ebola and marburg glycoprotein pseudovirions. Virology 2011, 419, 1–9. [Google Scholar]
- Bhattacharyya, S.; Warfield, K.L.; Ruthel, G.; Bavari, S.; Aman, M.J.; Hope, T.J. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology 2010, 401, 18–28. [Google Scholar]
- Sanchez, A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (b and l) activity. J. Infect. Dis. 2007, 196, S251–S258. [Google Scholar] [CrossRef]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204, S957–S967. [Google Scholar]
- Dolnik, O.; Kolesnikova, L.; Becker, S. Filoviruses: Interactions with the host cell. Cell Mol. Life Sci. 2008, 65, 756–776. [Google Scholar]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar]
- Cureton, D.K.; Massol, R.H.; Saffarian, S.; Kirchhausen, T.L.; Whelan, S.P. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009, 5. [Google Scholar]
- Rodriguez, N.E.; Gaur, U.; Wilson, M.E. Role of caveolae in leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell Microbiol. 2006, 8, 1106–1120. [Google Scholar]
- Quinn, K.; Brindley, M.A.; Weller, M.L.; Kaludov, N.; Kondratowicz, A.; Hunt, C.L.; Sinn, P.L.; McCray, P.B., Jr.; Stein, C.S.; Davidson, B.L.; et al. Rho gtpases modulate entry of ebola virus and vesicular stomatitis virus pseudotyped vectors. J. Virol. 2009, 83, 10176–10186. [Google Scholar]
- Cote, M.; Zheng, Y.M.; Liu, S.L. Receptor binding and low ph coactivate oncogenic retrovirus envelope-mediated fusion. J. Virol. 2009, 83, 11447–11455. [Google Scholar]
- Brindley, M.A.; Maury, W. Endocytosis and a low-ph step are required for productive entry of equine infectious anemia virus. J. Virol. 2005, 79, 14482–14488. [Google Scholar]
- Barnard, R.J.; Young, J.A. Alpharetrovirus envelope-receptor interactions. Curr. Top. Microbiol. Immunol. 2003, 281, 107–136. [Google Scholar]
- Wong, A.C.; Sandesara, R.G.; Mulherkar, N.; Whelan, S.P.; Chandran, K. A forward genetic strategy reveals destabilizing mutations in the ebolavirus glycoprotein that alter its protease dependence during cell entry. J. Virol. 2010, 84, 163–175. [Google Scholar]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin cleavage potentiates the ebola virus glycoprotein to undergo a subsequent fusion relevant conformational change. J. Virol. 2011. [Google Scholar]
- Martinez, O.; Johnson, J.; Manicassamy, B.; Rong, L.; Olinger, G.G.; Hensley, L.E.; Basler, C.F. Zaire ebola virus entry into human dendritic cells is insensitive to cathepsin l inhibition. Cell Microbiol. 2010, 12, 148–157. [Google Scholar]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal niemann-pick c1 is essential for ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter niemann-pick c1. Nature 2011, 477, 340–343. [Google Scholar]
- Rosenbaum, A.I.; Maxfield, F.R. Niemann-pick type c disease: Molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011, 116, 789–795. [Google Scholar]
- Poh, M.K.; Shui, G.; Xie, X.; Shi, P.Y.; Wenk, M.R.; Gu, F. U18666a, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antivir. Res. 2012, 93, 191–198. [Google Scholar]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Lai, M.M. Hepatitis c virus egress and release depend on endosomal trafficking of core protein. J. Virol. 2010, 84, 11590–11598. [Google Scholar]
- Hagiwara, K.; Nakamura, Y.; Nishijima, M.; Yamakawa, Y. Prevention of prion propagation by dehydrocholesterol reductase inhibitors in cultured cells and a therapeutic trial in mice. Biol. Pharm. Bull. 2007, 30, 835–838. [Google Scholar]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the niemann-pick c1-like 1 cholesterol absorption receptor as a new hepatitis c virus entry factor. Nat. Med. 2012. [Google Scholar]
- Gregory, S.M.; Harada, E.; Liang, B.; Delos, S.E.; White, J.M.; Tamm, L.K. Structure and function of the complete internal fusion loop from ebolavirus glycoprotein 2. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11211–11216. [Google Scholar]
- Bale, S.; Liu, T.; Li, S.; Wang, Y.; Abelson, D.; Fusco, M.; Woods, V.L., Jr.; Ollmann Saphire, E. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar]
- Freitas, M.S.; Gaspar, L.P.; Lorenzoni, M.; Almeida, F.C.; Tinoco, L.W.; Almeida, M.S.; Maia, L.F.; Degreve, L.; Valente, A.P.; Silva, J.L. Structure of the ebola fusion peptide in a membrane-mimetic environment and the interaction with lipid rafts. J. Biol. Chem. 2007, 282, 27306–27314. [Google Scholar]
- Gomara, M.J.; Mora, P.; Mingarro, I.; Nieva, J.L. Roles of a conserved proline in the internal fusion peptide of ebola glycoprotein. FEBS Lett. 2004, 569, 261–266. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hunt, C.L.; Lennemann, N.J.; Maury, W. Filovirus Entry: A Novelty in the Viral Fusion World. Viruses 2012, 4, 258-275. https://doi.org/10.3390/v4020258
Hunt CL, Lennemann NJ, Maury W. Filovirus Entry: A Novelty in the Viral Fusion World. Viruses. 2012; 4(2):258-275. https://doi.org/10.3390/v4020258
Chicago/Turabian StyleHunt, Catherine L., Nicholas J. Lennemann, and Wendy Maury. 2012. "Filovirus Entry: A Novelty in the Viral Fusion World" Viruses 4, no. 2: 258-275. https://doi.org/10.3390/v4020258
APA StyleHunt, C. L., Lennemann, N. J., & Maury, W. (2012). Filovirus Entry: A Novelty in the Viral Fusion World. Viruses, 4(2), 258-275. https://doi.org/10.3390/v4020258