Arenavirus Evasion of Host Anti-Viral Responses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune Response to the Old World Arenaviruses
2.1. Type I Interferon Responses
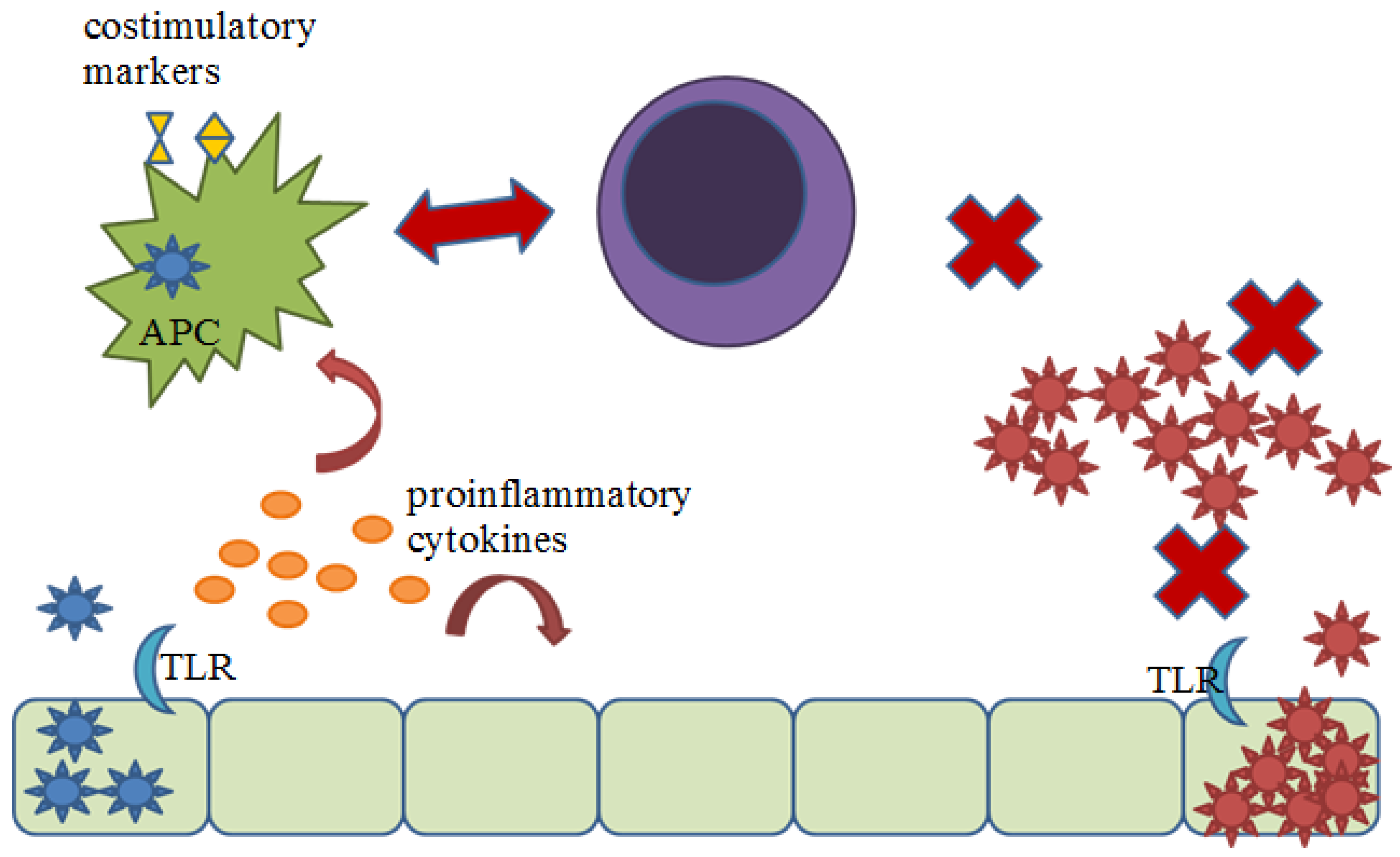
2.2. Proinflammatory Responses

2.3. Adaptive Immune Response
3. Immune Evasion
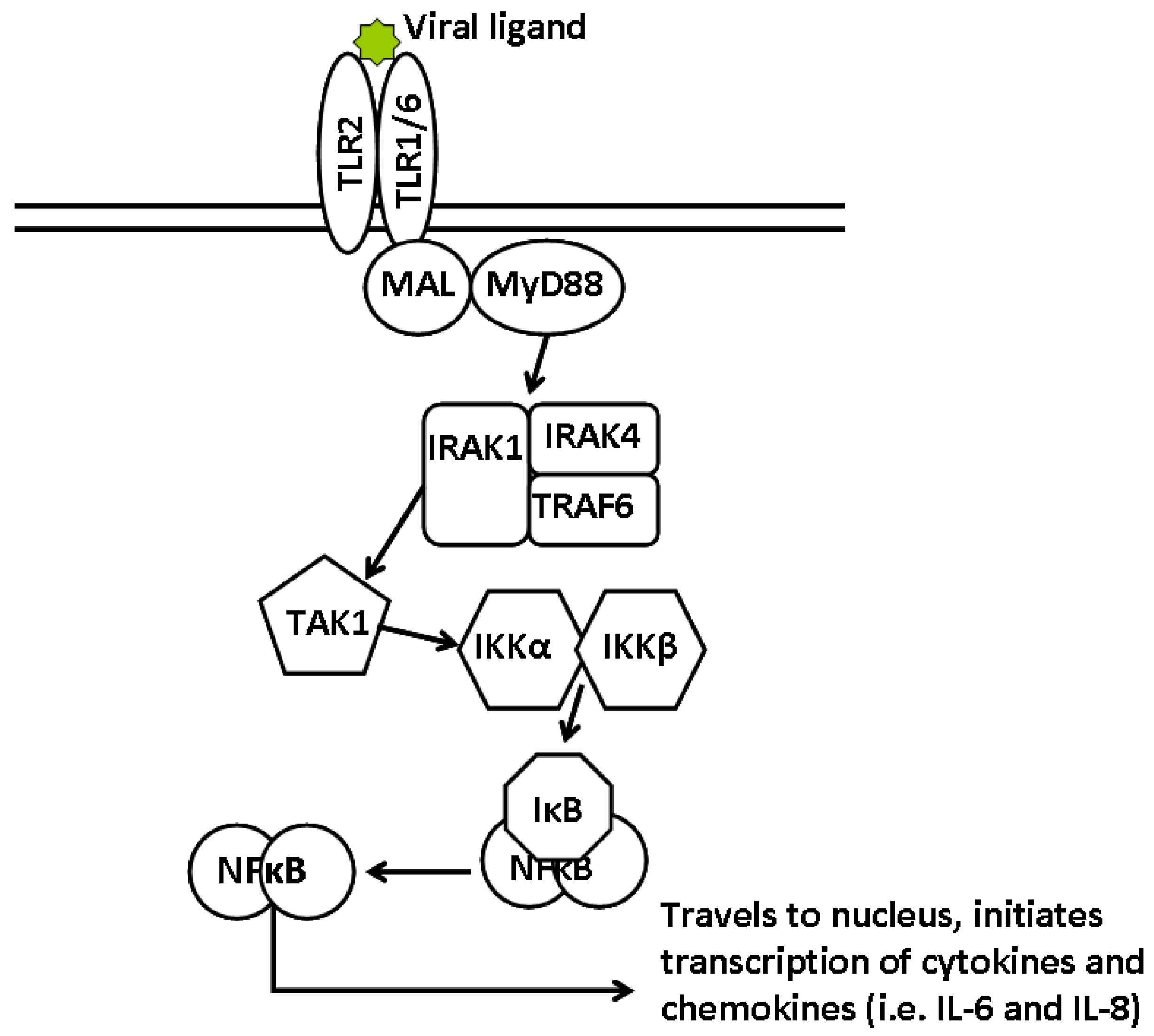
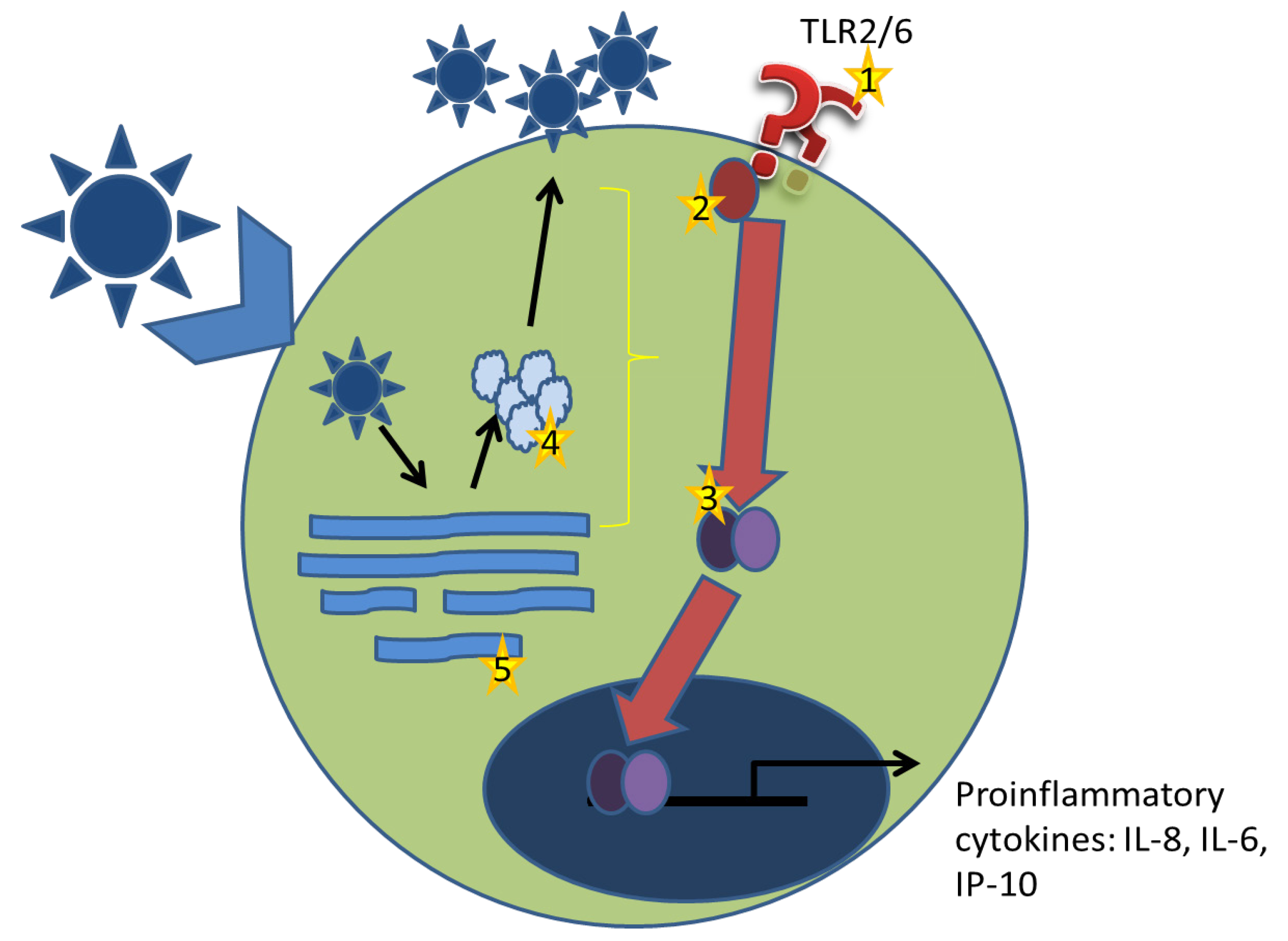
4. Arenaviruses and TLR2
4.1. Inhibition of the Proinflammatory Response

5. Future Directions


Conflict of Interest
Supplementary Files
References
- Biron, C.A.; Nguyen, K.B.; Pien, G.C. Innate immune responses to LCMV infections: Natural killer cells and cytokines. Curr. Top. Microbiol. Immunol. 2002, 263, 7–27. [Google Scholar]
- Merigan, T.C.; Oldstone, M.B.; Welsh, R.M. Interferon production during lymphocytic choriomeningitis virus infection of nude and normal mice. Nature 1977, 268, 67–68. [Google Scholar] [CrossRef]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C.; Fei, Z.; Folkerts, O.; Sobral, B.; Swindells, M.; Bryant, J.; Davis, H.; Pauza, C.D.; et al. Early blood profiles of virus infection in a monkey model for lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar]
- Rodas, J.D.; Cairo, C.; Djavani, M.; Zapata, J.C.; Ruckwardt, T.; Bryant, J.; Pauza, C.D.; Lukashevich, I.S.; Salvato, M.S. Circulating natural killer and gammadelta t cells decrease soon after infection of rhesus macaques with lymphocytic choriomeningitis virus. Mem. Inst. Oswaldo. Cruz. 2009, 104, 583–591. [Google Scholar] [CrossRef]
- Asper, M.; Sternsdorf, T.; Hass, M.; Drosten, C.; Rhode, A.; Schmitz, H.; Gunther, S. Inhibition of different lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic arenavirus. J. Virol. 2004, 78, 3162–3169. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; et al. Processing of genome 5' termini as a strategy of negative-strand rna viruses to avoid rig-i-dependent interferon induction. PLoS One 2008, 3, e2032. [Google Scholar]
- Pannetier, D.; Faure, C.; Georges-Courbot, M.C.; Deubel, V.; Baize, S. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to mopeia virus infection. J. Virol. 2004, 78, 10516–10524. [Google Scholar] [CrossRef]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of lassa and marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type i interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef]
- Jung, A.; Kato, H.; Kumagai, Y.; Kumar, H.; Kawai, T.; Takeuchi, O.; Akira, S. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic t-cell response via myd88. J. Virol. 2008, 82, 196–206. [Google Scholar] [CrossRef]
- Camus, G.S.; Qui, X.; Bente, D.A.; Strong, U.C.; Jones, S.M. Resistance to a Lassa virus infection is interferon-dependent, but not B or T cell-mediated in mice. Abstracts for the 28th Meeting of the American Society for Virology 2009, W7-7, 94. [Google Scholar]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Ben-Baruch, A.; Michiel, D.F.; Oppenheim, J.J. Signals and receptors involved in recruitment of inflammatory cells. J. Biol. Chem. 1995, 270, 11703–11706. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevich, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on il-8 and tnf-alpha gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Fennewald, S.M.; Aronson, J.F.; Zhang, L.; Herzog, N.K. Alterations in nf-kappab and rbp-jkappa by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 2002, 76, 1154–1162. [Google Scholar] [CrossRef]
- Fennewald, S.M.; Scott, E.P.; Zhang, L.; Yang, X.; Aronson, J.F.; Gorenstein, D.G.; Luxon, B.A.; Shope, R.E.; Beasley, D.W.; Barrett, A.D.; et al. Thioaptamer decoy targeting of ap-1 proteins influences cytokine expression and the outcome of arenavirus infections. J. Gen. Virol. 2007, 88, 981–990. [Google Scholar] [CrossRef]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. Myd88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef]
- Barbalat, R.; Lau, L.; Locksley, R.M.; Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type i interferon in response to viral but not bacterial ligands. Nat. Immunol. 2009, 10, 1200–1207. [Google Scholar] [CrossRef]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of cns glial cells results in tlr2-myd88/mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic old world arenaviruses inhibit tlr2/mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; McCormick, J.B. Towards a human lassa fever vaccine. Rev. Med. Virol. 2001, 11, 331–341. [Google Scholar] [CrossRef]
- ter Meulen, J.; Badusche, M.; Kuhnt, K.; Doetze, A.; Satoguina, J.; Marti, T.; Loeliger, C.; Koulemou, K.; Koivogui, L.; Schmitz, H.; et al. Characterization of human cd4(+) t-cell clones recognizing conserved and variable epitopes of the lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar] [CrossRef]
- Meulen, J.; Badusche, M.; Satoguina, J.; Strecker, T.; Lenz, O.; Loeliger, C.; Sakho, M.; Koulemou, K.; Koivogui, L.; Hoerauf, A. Old and new world arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by lassa virus-specific human cd4+ t-cell clones. Virology 2004, 321, 134–143. [Google Scholar] [CrossRef]
- Zhou, S.; Kurt-Jones, E.A.; Cerny, A.M.; Chan, M.; Bronson, R.T.; Finberg, R.W. Myd88 intrinsically regulates cd4 t-cell responses. J. Virol. 2009, 83, 1625–1634. [Google Scholar] [CrossRef]
- Bartholdy, C.; Christensen, J.E.; Grujic, M.; Christensen, J.P.; Thomsen, A.R. T-cell intrinsic expression of myd88 is required for sustained expansion of the virus-specific cd8+ t-cell population in lcmv-infected mice. J. Gen. Virol. 2009, 90, 423–431. [Google Scholar] [CrossRef]
- Rahman, A.H.; Cui, W.; Larosa, D.F.; Taylor, D.K.; Zhang, J.; Goldstein, D.R.; Wherry, E.J.; Kaech, S.M.; Turka, L.A. Myd88 plays a critical t cell-intrinsic role in supporting cd8 t cell expansion during acute lymphocytic choriomeningitis virus infection. J. Immunol. 2008, 181, 3804–3810. [Google Scholar]
- Rahman, A.H.; Zhang, R.; Blosser, C.D.; Hou, B.; Defranco, A.L.; Maltzman, J.S.; Wherry, E.J.; Turka, L.A. Antiviral memory cd8 t-cell differentiation, maintenance, and secondary expansion occur independently of myd8. Blood 2011, 117, 3123–3130. [Google Scholar] [CrossRef]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliott, L.H.; King, I.J. Clinical virology of lassa fever in hospitalized patients. J. Infect. Dis. 1987, 155, 456–464. [Google Scholar] [CrossRef]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Moraz, M.L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert. Rev. Anti Infect. Ther. 2011, 9, 49–59. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Tikhonov, I.; Rodas, J.D.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. Arenavirus-mediated liver pathology: Acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, II; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. Lcmv-mediated hepatitis in rhesus macaques: We but not arm strain activates hepatocytes and induces liver regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar] [CrossRef]
- Zapata, J.; While, D.; Poonia, B.; Mani, S.; Jett, M.; Carrion, R., Jr.; Crasta, O.; Zhang, Y.; Salvato, M.; Lukashevich, I. Expression of coagulation factor thrombomodulin is increased in cells exposed to lassa virus. In Proceedings of The 29th ASV Meeting, Bozeman, Montana, USA, July 17, 2010.
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of lassa virus replication in human dendritic cells and macrophages. Microbes. Infect. 2006, 8, 1194–1202. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type i interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type i interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Pannetier, D.; Reynard, S.; Russier, M.; Journeaux, A.; Tordo, N.; Deubel, V.; Baize, S. Human dendritic cells infected with the nonpathogenic mopeia virus induce stronger t-cell responses than those infected with lassa virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de La Torre, J.C.; Oldstone, M.B. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in lassa virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar]
- Borrow, P.; Evans, C.F.; Oldstone, M.B. Virus-induced immunosuppression: Immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J. Virol. 1995, 69, 1059–1070. [Google Scholar]
- Sevilla, N.; McGavern, D.B.; Teng, C.; Kunz, S.; Oldstone, M.B. Viral targeting of hematopoietic progenitors and inhibition of dc maturation as a dual strategy for immune subversion. J. Clin. Inv. 2004, 113, 737–745. [Google Scholar]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; McRae, M.; Rollin, P.E.; Pulendran, B. Cutting edge: Impairment of dendritic cells and adaptive immunity by ebola and lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins b and h are necessary for tlr2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in tlr2-mediated macrophage activation by hepatitis c virus core and ns3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis c core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Sirisinha, S. Insight into the mechanisms regulating immune homeostasis in health and disease. Asian Pac. J. Allergy Immunol. 2011, 29, 1–14. [Google Scholar]
- Kirschning, C.J.; Dreher, S.; Maass, B.; Fichte, S.; Schade, J.; Koster, M.; Noack, A.; Lindenmaier, W.; Wagner, H.; Boldicke, T. Generation of anti-tlr2 intrabody mediating inhibition of macrophage surface tlr2 expression and tlr2-driven cell activation. BMC Biotechnol. 2010, 10, 31. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hayes, M.; Salvato, M. Arenavirus Evasion of Host Anti-Viral Responses. Viruses 2012, 4, 2182-2196. https://doi.org/10.3390/v4102182
Hayes M, Salvato M. Arenavirus Evasion of Host Anti-Viral Responses. Viruses. 2012; 4(10):2182-2196. https://doi.org/10.3390/v4102182
Chicago/Turabian StyleHayes, Melissa, and Maria Salvato. 2012. "Arenavirus Evasion of Host Anti-Viral Responses" Viruses 4, no. 10: 2182-2196. https://doi.org/10.3390/v4102182
APA StyleHayes, M., & Salvato, M. (2012). Arenavirus Evasion of Host Anti-Viral Responses. Viruses, 4(10), 2182-2196. https://doi.org/10.3390/v4102182