Junín Virus Pathogenesis and Virus Replication

{kind=link}
{kind=link}
Abstract
:1. Introduction
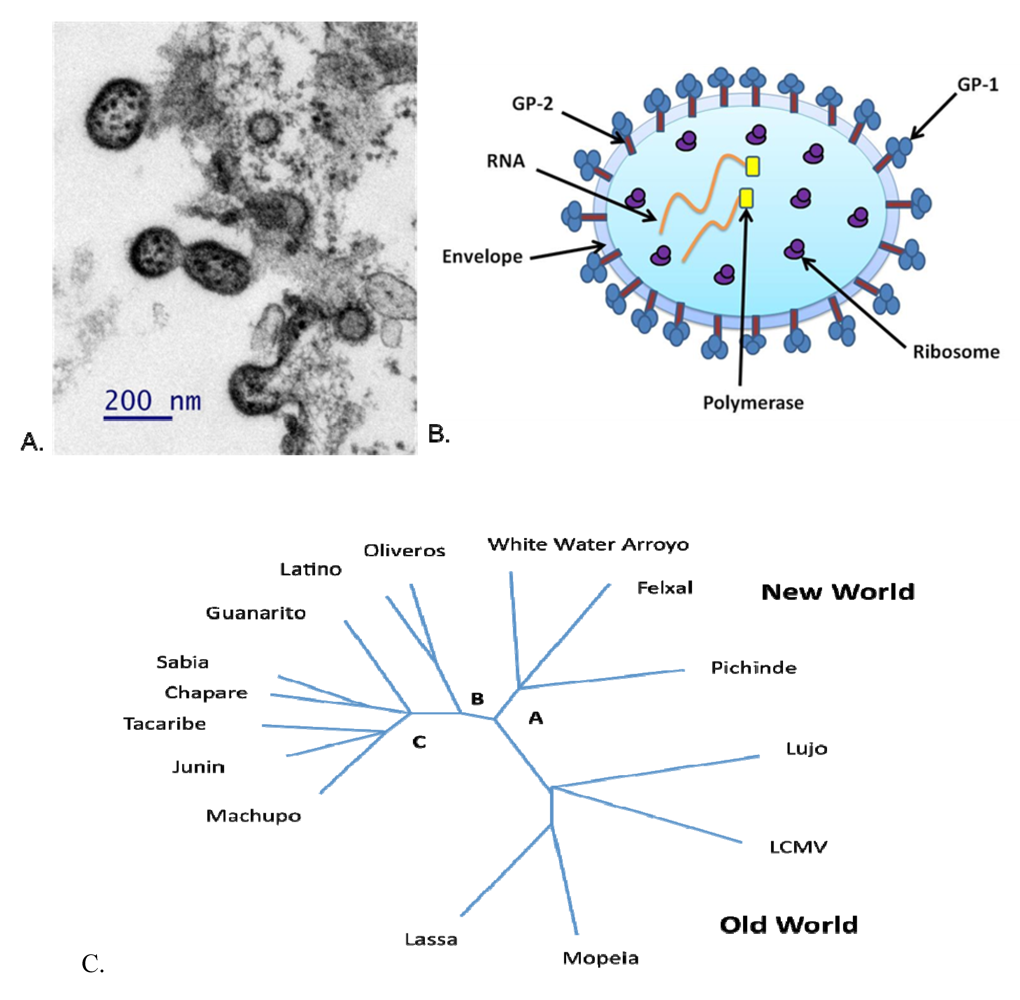
2. Agent

3. Epidemiology
4. Clinical Manifestations, Pathology and Pathogenesis
5. Animal Models
5.1 JUNV Infection in Mice
5.2. JUNV Infection in Guinea Pigs
5.3. JUNV Infection in Natural Host
5.4. Junín Infection in Non-human primates
6. Role of Adaptive Immune Response
7. Vascular dysfunction
8. Role of Glycoprotein in pathogenesis:
9. Target cells and cell entry
10. Innate Immune Response
11. Live-attenuated vaccines

12. Other vaccine preparations
13. Therapeutics
Conflict of Interest
References
- Mills, J.N.; Ellis, B.A.; Childs, J.E.; McKee, K.T., Jr.; Maiztegui, J.I.; Peters, C.J.; Ksiazek, T.G.; Jahrling, P.B. Prevalence of infection with junin virus in rodent populations in the epidemic area of argentine hemorrhagic fever. T. Am. J. Trop. Med. Hyg. 1994, 51, 554–562. [Google Scholar]
- Parodi, A.S.; Coto, C.E.; Boxaca, M.; Lajmanovich, S.; Gonzalez, S. Characteristics of junin virus. Etiological agent of argentine hemorrhagic fever. Archiv fur die gesamte Virusforschung 1966, 19, 393–402. [Google Scholar] [CrossRef]
- Buchmeier, M.J.d.l.T.J.; Peters, C.J. Arenaviridae: The viruses and their replication. In Fields virology; Knipe, D.M.H.P., Ed.; Wolter Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1791–1827. [Google Scholar]
- Tortorici, M.A.; Albarino, C.G.; Posik, D.M.; Ghiringhelli, P.D.; Lozano, M.E.; Rivera Pomar, R.; Romanowski, V. Arenavirus nucleocapsid protein displays a transcriptional antitermination activity in vivo. Vir. Res. 2001, 73, 41–55. [Google Scholar] [CrossRef]
- Meyer, B.J.; Southern, P.J. Sequence heterogeneity in the termini of lymphocytic choriomeningitis virus genomic and antigenomic rnas. J. Virol. 1994, 68, 7659–7664. [Google Scholar]
- Beyer, W.R.; Popplau, D.; Garten, W.; von Laer, D.; Lenz, O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase ski-1/s1p. J. Virol. 2003, 77, 2866–2872. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The lassa virus glycoprotein precursor gp-c is proteolytically processed by subtilase ski-1/s1p. P. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar]
- Pinschewer, D.D.; Perez, M.; Sanchez, A.B.; de la Torre, J.C. Recombinant lymphocytic choriomeningitis virus expressing vesicular stomatitis virus glycoprotein. P. Natl. Acad. Sci. USA 2003, 100, 7895–7900. [Google Scholar]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the rna-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small ring finger protein z drives arenavirus budding: Implications for antiviral strategies. P. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef]
- Strecker, T.; Eichler, R.; Meulen, J.; Weissenhorn, W.; Dieter Klenk, H.; Garten, W.; Lenz, O. Lassa virus z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for lassa virus budding. J. Virol. 2006, 80, 4191–4195. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of lujo virus, a new hemorrhagic fever-associated arenavirus from southern africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Enria, D.A.; Briggiler, A.M.; Pini, N.; Levis, S. Clinical manifestations of new world hantaviruses. Curr. Top. Microbiol. 2001, 256, 117–134. [Google Scholar]
- Briggiler, A.M.L.S.; Enriar, D.A.; Maiztegui, J.I. Argentine hemorrhagic fever in pregnant women. Medicina 1990, 50. [Google Scholar]
- McKee, K.T., Jr.; Mahlandt, B.G.; Maiztegui, J.I.; Green, D.E.; Peters, C.J. Virus-specific factors in experimental argentine hemorrhagic fever in rhesus macaques. J. Med. Virol. 1987, 22, 99–111. [Google Scholar] [CrossRef]
- Gonzalez, P.H.; Cossio, P.M.; Arana, R.; Maiztegui, J.I.; Laguens, R.P. Lymphatic tissue in argentine hemorrhagic fever. Pathologic features. Arch. Patho. l Lab. Med. 1980, 104, 250–254. [Google Scholar]
- Fennewald, S.M.; Aronson, J.F.; Zhang, L.; Herzog, N.K. Alterations in nf-kappab and rbp-jkappa by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 2002, 76, 1154–1162. [Google Scholar] [CrossRef]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Peters, C.J.; Liu, C.T.; Anderson, G.W., Jr.; Morrill, J.C.; Jahrling, P.B. Pathogenesis of viral hemorrhagic fevers: Rift valley fever and lassa fever contrasted. Rev Infect Dis 1989, 11 Suppl 4, S743–S749. [Google Scholar]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevich, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on il-8 and tnf-alpha gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Rambukkana, A.; Kunz, S.; Min, J.; Campbell, K.P.; Oldstone, M.B. Targeting schwann cells by nonlytic arenaviral infection selectively inhibits myelination. Proc. Natl. Acad. Sci. USA 2003, 100, 16071–16076. [Google Scholar]
- Enria, D.A.; Briggiler, A.M.; Fernandez, N.J.; Levis, S.C.; Maiztegui, J.I. Importance of dose of neutralising antibodies in treatment of argentine haemorrhagic fever with immune plasma. Lancet 1984, 2, 255–256. [Google Scholar]
- Peters, C.J.L.C.; Anderson, G.W.; Morrill, J.C., Jr; Jahrling, P.B. Pathogenesis of viral hemorrhagic fevers. Rev. Infect Dis. 1989, 11, S749–S749. [Google Scholar]
- Kolokoltsova, O.A.; Yun, N.E.; Poussard, A.L.; Smith, J.K.; Smith, J.N.; Salazar, M.; Walker, A.; Tseng, C.T.; Aronson, J.F.; Paessler, S. Mice lacking alpha/beta and gamma interferon receptors are susceptible to junin virus infection. J. Virol. 84, 13063–13067.
- Giovanniello, O.A.; Nejamkis, M.R.; Galassi, N.V.; Nota, N.R. Immunosuppression in experimental junin virus infection of mice. Intervirol. 1980, 13, 122–125. [Google Scholar] [CrossRef]
- Gomez, R.M.; Jaquenod de Giusti, C.; Sanchez Vallduvi, M.M.; Frik, J.; Ferrer, M.F.; Schattner, M. Junin virus. A xxi century update. Microbes and infection / Institut Pasteur 2011, 13, 303–311. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; de Guerrero, L.B.; Boxaca, M.C. Experimental biology and pathogenesis of junin virus infection in animals and man. B. World Health Organ. 1975, 52, 507–515. [Google Scholar]
- Campetella, O.E.; Galassi, N.V.; Sanjuan, N.; Barrios, H.A. Susceptible adult murine model for junin virus. J. Med. Virol. 1988, 26, 443–451. [Google Scholar] [CrossRef]
- Campetella, O.E.; Barrios, H.A.; Galassi, N.V. Junin virus-induced delayed-type hypersensitivity suppression in adult mice. J. Med. Virol. 1988, 25, 227–235. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Calello, M.A.; Quintans, C.J.; Panisse, H.; Woyskowsky, N.M.; Zannoli, V.H. Junin virus infection in genetically athymic mice. Intervirol. 1983, 19, 1–5. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Schmunis, G.A.; Parodi, A.S. Junin virus multiplication in thymectomized mice. Effect of thymus and immunocompetent cells grafting. Arch. Gesamte Virusforsch. 1969, 26, 63–73. [Google Scholar] [CrossRef]
- Budzko, D.B.; Casals, J.; Waksman, B.H. Enhanced resistance against junin virus infection induced by corynebacterium parvum. Infect. Immun. 1978, 19, 893–897. [Google Scholar]
- Nejamkis, M.R.; Nota, N.R.; Weissenbacher, M.C.; Guerrero, L.B.; Giovanniello, O.A. Passive immunity against junin virus in mice. Acta Virol. 1975, 19, 237–244. [Google Scholar]
- Nota, N.R.; Nejamkis, M.R.; Giovanniello, O.A. Further experiments on the action of antithymocyte serum in experimental junin virus infection. Acta Virol. 1976, 20, 61–65. [Google Scholar]
- Rabinovich, R.D.; Calello, M.A.; Boxaca, M.C.; Quintans, C.J.; Weissenbacher, M.C. Mouse splenocyte transfer effect depends on donor's junin virus infection stage. Intervirol. 1988, 29, 21–26. [Google Scholar] [CrossRef]
- Campetella, O.E.; Galassi, N.V.; Barrios, H.A. Contrasuppressor cells induced by junin virus. Immunology 1990, 69, 629–631. [Google Scholar]
- Kenyon, R.H.; Green, D.E.; Peters, C.J. Effect of immunosuppression on experimental argentine hemorrhagic fever in guinea pigs. J. Virol. 1985, 53, 75–80. [Google Scholar]
- Carballal, G.; Cossio, P.M.; Laguens, R.P.; Ponzinibbio, C.; Oubina, J.R.; Meckert, P.C.; Rabinovich, A.; Arana, R.M. Junin virus infection of guinea pigs: Immunohistochemical and ultrastructural studies of hemopoietic tissue. J. Infect. Dis. 1981, 143, 7–14. [Google Scholar] [CrossRef]
- Elsner, B.; Boxaca, M.C.; Weissenbacher, M.; De Guerrero, L.B. [experimental junin virus infection of guinea pigs. I. Pathological anatomy]. Medicina (B Aires) 1976, 36, 197–206. [Google Scholar]
- Maiztegui, J.I.; Laguens, R.P.; Cossio, P.M.; Casanova, M.B.; de la Vega, M.T.; Ritacco, V.; Segal, A.; Fernandez, N.J.; Arana, R.M. Ultrastructural and immunohistochemical studies in five cases of argentine hemorrhagic fever. J. Infect. Dis. 1975, 132, 35–53. [Google Scholar] [CrossRef]
- Kenyon, R.H.; Green, D.E.; Maiztegui, J.I.; Peters, C.J. Viral strain dependent differences in experimental argentine hemorrhagic fever (junin virus) infection of guinea pigs. Intervirol. 1988, 29, 133–143. [Google Scholar]
- Yun, N.E.; Linde, N.S.; Dziuba, N.; Zacks, M.A.; Smith, J.N.; Smith, J.K.; Aronson, J.F.; Chumakova, O.V.; Lander, H.M.; Peters, C.J.; et al. Pathogenesis of xj and romero strains of junin virus in two strains of guinea pigs. Am. J. Trop. Med. Hyg. 2008, 79, 275–282. [Google Scholar]
- Oubina, J.R.; Carballal, G. Neurotropism of a high-passage xj strain of junin virus. J. Med. Virol. 1985, 15, 157–161. [Google Scholar] [CrossRef]
- Laguens, R.M.; Avila, M.M.; Samoilovich, S.R.; Weissenbacher, M.C.; Laguens, R.P. Pathogenicity of an attenuated strain (xjcl3) of junin virus. Morphological and virological studies in experimentally infected guinea pigs. Intervirol. 1983, 20, 195–201. [Google Scholar] [CrossRef]
- Sabattini, M.S.; Maiztegui, J.I. [argentine hemorrhagic fever]. Medicina (B Aires) 1970, 30 Suppl 1, 111–128. [Google Scholar]
- Vitullo, A.D.; Hodara, V.L.; Merani, M.S. Effect of persistent infection with junin virus on growth and reproduction of its natural reservoir, calomys musculinus. Am. J. Trop. Med. Hyg. 1987, 37, 663–669. [Google Scholar]
- Lampuri, J.S.; Vidal, M.D.; Coto, C.E. [response of calomys musculinus to experimental infection with junin virus]. Medicina 1982, 42, 61–66. [Google Scholar]
- Sabattini, M.S.; Gonzalez, L.E.; de Rios Diaz, G.; Vega, V.R. Infeccion natural y experimental de roedores con virus junin. Medicina (B Aires) 1977, 37, 149–161. [Google Scholar]
- Chiappero, M.B.; Gardenal, C.N. Restricted gene flow in calomys musculinus (rodentia, muridae), the natural reservoir of junin virus. J. Hered. 2003, 94, 490–495. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Calello, M.A.; Colillas, O.J.; Rondinone, S.N.; Frigerio, M.J. Argentine hemorrhagic fever: A primate model. Intervirol. 1979, 11, 363–365. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Avila, M.M.; Calello, M.A.; Merani, M.S.; McCormick, J.B.; Rodriguez, M. Effect of ribavirin and immune serum on junin virus-infected primates. Med. Microbiol. Immunol. 1986, 175, 183–186. [Google Scholar]
- Weissenbacher, M.C.; Coto, C.E.; Calello, M.A.; Rondinone, S.N.; Damonte, E.B.; Frigerio, M.J. Cross-protection in nonhuman primates against argentine hemorrhagic fever. Infect. Immun. 1982, 35, 425–430. [Google Scholar]
- Samoilovich, S.R.; Calello, M.A.; Laguens, R.P.; Weissenbacher, M.C. Long-term protection against argentine hemorrhagic fever in tacaribe virus infected marmosets: Virologic and histopathologic findings. J. Med. Virol. 1988, 24, 229–236. [Google Scholar] [CrossRef]
- Samoilovich, S.R.; Pecci Saavedra, J.; Frigerio, M.J.; Weissenbacher, M.C. Nasal and intrathalamic inoculations of primates with tacaribe virus: Protection against argentine hemorrhagic fever and absence of neurovirulence. Acta Virol. 1984, 28, 277–281. [Google Scholar]
- McKee, K.T., Jr.; Oro, J.G.; Kuehne, A.I.; Spisso, J.A.; Mahlandt, B.G. Safety and immunogenicity of a live-attenuated junin (argentine hemorrhagic fever) vaccine in rhesus macaques. Am. J. Trop. Med. Hyg. 1993, 48, 403–411. [Google Scholar]
- McKee, K.T., Jr.; Oro, J.G.; Kuehne, A.I.; Spisso, J.A.; Mahlandt, B.G. Candid no. 1 argentine hemorrhagic fever vaccine protects against lethal junin virus challenge in rhesus macaques. Intervirol. 1992, 34, 154–163. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Hesse, H.R.; Eddy, G.A.; Johnson, K.M.; Callis, R.T.; Stephen, E.L. Lassa virus infection of rhesus monkeys. J. Infect. Dis. 1980, 141, 580–589. [Google Scholar] [CrossRef]
- Vallejos, D.A.; Ambrosio, A.M.; Feuillade, M.R.; Maiztegui, J.I. Lymphocyte subsets alteration in patients with argentine hemorrhagic fever. J. Med. Virol. 1989, 27, 160–163. [Google Scholar] [CrossRef]
- Ambrosio, A.M.; Gamboa, G.S.; Feuillade, M.R.; Briggiler, A.M.; Rimoldi, M.T. Peripheral blood leukocytes of patients with argentine hemorrhagic fever as effectors of antibody-dependent cell-cytotoxicity. J. Med. Virol. 1992, 37, 232–236. [Google Scholar] [CrossRef]
- Emonet, S.F.; Seregin, A.V.; Yun, N.E.; Poussard, A.L.; Walker, A.G.; de la Torre, J.C.; Paessler, S. Rescue from cloned cdnas and in vivo characterization of recombinant pathogenic romero and live-attenuated candid #1 strains of junin virus, the causative agent of argentine hemorrhagic fever disease. J. Virol. 2011, 85, 1473–1483. [Google Scholar]
- Avila, M.M.; Samoilovich, S.R.; Laguens, R.P.; Merani, M.S.; Weissenbacher, M.C. Protection of junin virus-infected marmosets by passive administration of immune serum: Association with late neurologic signs. J. Med. Virol. 1987, 21, 67–74. [Google Scholar] [CrossRef]
- Enria, D.A.; Briggiler, A.M.; Sanchez, Z. Treatment of argentine hemorrhagic fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Kunz, S. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb. Haemostasis. 2009, 102, 1024–1029. [Google Scholar]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef]
- Andrews, B.S.; Theofilopoulos, A.N.; Peters, C.J.; Loskutoff, D.J.; Brandt, W.E.; Dixon, F.J. Replication of dengue and junin viruses in cultured rabbit and human endothelial cells. Infect. Immun. 1978, 20, 776–781. [Google Scholar]
- Gomez, R.M.; Pozner, R.G.; Lazzari, M.A.; D'Atri, L.P.; Negrotto, S.; Chudzinski-Tavassi, A.M.; Berria, M.I.; Schattner, M. Endothelial cell function alteration after junin virus infection. Thromb. Haemostasis. 2003, 90, 326–333. [Google Scholar]
- Elsner, B.; Schwarz, E.; Mando, O.G.; Maiztegui, J.; Vilches, A. Pathology of 12 fatal cases of argentine hemorrhagic fever. T. Am. J. Trop. Med. Hyg. 1973, 22, 229–236. [Google Scholar]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for new world haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Kuhn, J.H.; Spiropoulou, C.F.; Albarino, C.G.; Nguyen, D.P.; Salazar-Bravo, J.; Dorfman, T.; Lee, A.S.; Wang, E.; Ross, S.R.; et al. Receptor determinants of zoonotic transmission of new world hemorrhagic fever arenaviruses. P. Natl. Acad. Sci. USA 2008, 105, 2664–2669. [Google Scholar]
- Abraham, J.; Corbett, K.D.; Farzan, M.; Choe, H.; Harrison, S.C. Structural basis for receptor recognition by new world hemorrhagic fever arenaviruses. Nat. Ctruct. Mol. Biol. 2010, 17, 438–444. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Exline, C.M.; Rathbun, J.Y.; Cannon, P.M. Substitutions in the glycoprotein (gp) of the candid#1 vaccine strain of junin virus increase dependence on human transferrin receptor 1 for entry and destabilize the metastable conformation of gp. J. Virol. 2011, 85, 13457–13462. [Google Scholar] [CrossRef]
- Albarino, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Flint, M.; Bergeron, E.; White, D.M.; Nichol, S.T. The major determinant of attenuation in mice of the candid1 vaccine for argentine hemorrhagic fever is located in the g2 glycoprotein transmembrane domain. J. Virol. 2011, 85, 10404–10408. [Google Scholar]
- Ambrosio, A.M.; Enria, D.A.; Maiztegui, J.I. Junin virus isolation from lympho-mononuclear cells of patients with argentine hemorrhagic fever. Intervirol. 1986, 25, 97–102. [Google Scholar] [CrossRef]
- Ambrosio, M.; Vallejos, A.; Saavedra, C.; Maiztegui, J.I. Junin virus replication in peripheral blood mononuclear cells of patients with argentine haemorrhagic fever. Acta Virol. 1990, 34, 58–63. [Google Scholar]
- Laguens, M.; Chambo, J.G.; Laguens, R.P. In vivo replication of pathogenic and attenuated strains of junin virus in different cell populations of lymphatic tissue. Infect. Immun. 1983, 41, 1279–1283. [Google Scholar]
- Laguens, R.M.; Chambo, J.G.; Laguens, R.P. Splenic dendritic cells and junin virus. Med. Microbiol. Immun. 1986, 175, 187–189. [Google Scholar] [CrossRef]
- Medeot, S.I.; Contigiani, M.S.; Sabattini, M.S.; Camara, A. The role of mononuclear blood cells in experimental junin virus spread to the central nervous system. Vir. Immun. y 1995, 8, 101–108. [Google Scholar] [CrossRef]
- Alche, L.E.; Coulombie, F.C.; Coto, C.E. [isolation of junin virus from blood and peripheral lymphocytes of infected calomys musculinus]. Rev. Argent. Microbiol. 1985, 17, 177–181. [Google Scholar]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Neglect. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef]
- Blejer, J.L.; Remesar, M.C.; Mosca, E.E.; Nejamkis, M.R. [role of macrophages in the dissemination of the junin virus in the rat]. Rev. Argent. Microbiol. 1985, 17, 229–232. [Google Scholar]
- Coulombie, F.C.; Alche, L.E.; Lampuri, J.S.; Coto, C.E. Role of calomys musculinus peritoneal macrophages in age-related resistance to junin virus infection. J. Med. Virol. 1986, 18, 289–298. [Google Scholar] [CrossRef]
- Blejer, J.L.; Remesar, M.C.; Lerman, G.D.; Nejamkis, M.R. Macrophage maturity and modulation of response to junin virus in infected rats. J. Infect. Dis. 1986, 154, 478–482. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Correlation between endogenous interferon and the clinical evolution of patients with argentine hemorrhagic fever. J. Interfer. Res. 1985, 5, 383–389. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Falcoff, E.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Endogenous interferon in argentine hemorrhagic fever. J. Infect. Dis. 1984, 149, 428–433. [Google Scholar] [CrossRef]
- Lerer, G.D.; Saavedra, M.C.; Falcoff, R.; Maiztegui, J.I.; Molinas, F.C. Activity of a platelet protein kinase that phosphorylates fibrinogen and histone in argentine hemorrhagic fever. Acta Physiol. Pharm. L. 1991, 41, 377–386. [Google Scholar]
- Pozner, R.G.; Ure, A.E.; Jaquenod de Giusti, C.; D'Atri, L.P.; Italiano, J.E.; Torres, O.; Romanowski, V.; Schattner, M.; Gomez, R.M. Junin virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type i ifn signaling. PLoS Pathog. 2010, 6, e1000847. [Google Scholar]
- Dejean, C.B.; Ayerra, B.L.; Teyssie, A.R. Interferon response in the guinea pig infected with junin virus. J. Med. Virol. 1987, 23, 83–91. [Google Scholar] [CrossRef]
- Kenyon, R.H.; McKee, K.T., Jr.; Zack, P.M.; Rippy, M.K.; Vogel, A.P.; York, C.; Meegan, J.; Crabbs, C.; Peters, C.J. Aerosol infection of rhesus macaques with junin virus. Intervirol. 1992, 33, 23–31. [Google Scholar]
- Marta, R.F.; Montero, V.S.; Hack, C.E.; Sturk, A.; Maiztegui, J.I.; Molinas, F.C. Proinflammatory cytokines and elastase-alpha-1-antitrypsin in argentine hemorrhagic fever. T. Am. J. Trop. Med. Hyg. 1999, 60, 85–89. [Google Scholar]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martinez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase ikk{varepsilon}. J. Virol. 2012. [Google Scholar]
- Rodrigo, W.W.; Ortiz-Riano, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martinez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa b. J. Virol. 2012. [Google Scholar]
- Ortiz-Riano, E.; Cheng, B.Y.; de la Torre, J.C.; Martinez-Sobrido, L. The c-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in np-z interaction and counteraction of the type i interferon response. J. Virol. 2011, 85, 13038–13048. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type i interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type i interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of new world arenaviruses bind rig-i and interfere with type i interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; de la Torre, J.C.; et al. Junin virus infection activates the type i interferon pathway in a rig-i-dependent manner. PLoS Neglect. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- de Holstein, B.A.; Knecher, M.; Teyssie, A.R. [ini vitro interferon induction by junin virus; effect of pre-treatment with the inhibitor]. REv. Asoc. Argent. Microbiol. 1977, 9, 22–27. [Google Scholar]
- Remesar, M.C.; Blejer, J.L.; Lerman, G.D.; Dejean, C.; Nejamkis, M.R. [protection against encephalitis in rats caused by a pathogenic strain of the junin virus, using peripheral inoculation of an attenuated strain]. Rev. Argent. Microbiol. 1989, 21, 120–126. [Google Scholar]
- Kolokoltsova, O.A.; Yun, N.E.; Poussard, A.L.; Smith, J.K.; Smith, J.N.; Salazar, M.; Walker, A.; Tseng, C.T.; Aronson, J.F.; Paessler, S. Mice lacking alpha/beta and gamma interferon receptors are susceptible to junin virus infection. J. Virol. 2010, 84, 13063–13067. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Coto, C.E.; Calello, M.A. Cross-protection between tacaribe complex viruses. Presence of neutralizing antibodies against junin virus (argentine hemorrhagic fever) in guinea pigs infected with tacaribe virus. Intervirol. 1975, 6, 42–49. [Google Scholar] [CrossRef]
- Carballal, G.; Calello, M.A.; Laguens, R.P.; Weissenbacher, M.C. Tacaribe virus: A new alternative for argentine hemorrhagic fever vaccine. J. Med. Virol. 1987, 23, 257–263. [Google Scholar] [CrossRef]
- Parodi, A.S.; Greenway, D.J.; Rugiero, N.R.; Rivero, E.; Frigerio, M.J.; Mettler, W.E.; Garzon, F.; Boxaca, M.; Guerrero, L.B.; Nota, N.R. Sobre la etiologia del bute epidemico de junin. Dia Medico 1958, 30, 2300–2302. [Google Scholar]
- Candurra, N.A.; Damonte, E.B.; Coto, C.E. Antigenic relationships between attenuated and pathogenic strains of junin virus. J. Med. Virol. 1989, 27, 145–150. [Google Scholar] [CrossRef]
- Ruggiero, H.A.; Perez Isquierdo, F.; Milani, H.A.; Barri, A.; Val, A.; Maglio, F.; Astarloa, L.; Gonzalez Cambaceres, C.; Milani, H.L.; Tallone, J.C. [treatment of argentine hemorrhagic fever with convalescent's plasma. 4433 cases]. Presse Med 1986, 15, 2239–2242. [Google Scholar]
- de Guerrero, L.B.; Boxaca, M.C.; Malumbres, E.; Dejean, C.; Caruso, E. Early protection to junin virus of guinea pig with an attenuated junin virus strain. Acta Virol. 1985, 29, 334–337. [Google Scholar]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Hum. Vaccines 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Albarino, C.G.; Ghiringhelli, P.D.; Posik, D.M.; Lozano, M.E.; Ambrosio, A.M.; Sanchez, A.; Romanowski, V. Molecular characterization of attenuated junin virus strains. J. Gen. Virol. 1997, 78 ( Pt 7), 1605–1610. [Google Scholar]
- Medeot, S.I.; Contigiani, M.S.; Brandan, E.R.; Sabattini, M.S. Neurovirulence of wild and laboratory junin virus strains in animal hosts. J. Med. Virol. 1990, 32, 171–182. [Google Scholar] [CrossRef]
- Maiztegui, J.I.; McKee, K.T. , Jr.; Barrera Oro, J.G.; Harrison, L.H.; Gibbs, P.H.; Feuillade, M.R.; Enria, D.A.; Briggiler, A.M.; Levis, S.C.; Ambrosio, A.M.; et al. Protective efficacy of a live attenuated vaccine against argentine hemorrhagic fever. Ahf study group. J. Infect. Dis. 1998, 177, 277–283. [Google Scholar]
- Cresta, B.; Padula, P.; de Martinez Segovia, M. Biological properties of junin virus proteins. I. Identification of the immunogenic glycoprotein. Intervirol. 1980, 13, 284–288. [Google Scholar] [CrossRef]
- Videla, C.; Carballal, G.; Remorini, P.; La Torre, J. Formalin inactivated junin virus: Immunogenicity and protection assays. J. Med. Virol. 1989, 29, 215–220. [Google Scholar] [CrossRef]
- Seregin, A.V.; Yun, N.E.; Poussard, A.L.; Peng, B.H.; Smith, J.K.; Smith, J.N.; Salazar, M.; Paessler, S. Tc83 replicon vectored vaccine provides protection against junin virus in guinea pigs. Vaccine 2010, 28, 4713–4718. [Google Scholar]
- Maiztegui, J.I.; Fernandez, N.J.; de Damilano, A.J. Efficacy of immune plasma in treatment of argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet 1979, 2, 1216–1217. [Google Scholar]
- Kenyon, R.H.; Condie, R.M.; Jahrling, P.B.; Peters, C.J. Protection of guinea pigs against experimental argentine hemorrhagic fever by purified human igg: Importance of elimination of infected cells. Microbiol. Pathogen. 1990, 9, 219–226. [Google Scholar] [CrossRef]
- Parker, W.B. Metabolism and antiviral activity of ribavirin. Vir. Res. 2005, 107, 165–171. [Google Scholar] [CrossRef]
- Stephen, E.L.; Jones, D.E.; Peters, C.J.; Eddy, G.A.; Loizeaux, P.S.; Jahrling, P.B. Ribavirin, a broad spectrum antiviral agent. In Ribavirin treatment of toga-, areana- and bunyavirus infections in subhuman primates and other laboratory animals species; Academic Press: New York, NY, USA, 1980. [Google Scholar]
- McKee, K.T., Jr.; Huggins, J.W.; Trahan, C.J.; Mahlandt, B.G. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob. Agents Ch. 1988, 32, 1304–1309. [Google Scholar] [CrossRef]
- Barry, M.; Russi, M.; Armstrong, L.; Geller, D.; Tesh, R.; Dembry, L.; Gonzalez, J.P.; Khan, A.S.; Peters, C.J. Brief report: Treatment of a laboratory-acquired sabia virus infection. New Engl. J. Med. 1995, 333, 294–296. [Google Scholar] [CrossRef]
- Kilgore, P.E.; Ksiazek, T.G.; Rollin, P.E.; Mills, J.N.; Villagra, M.R.; Montenegro, M.J.; Costales, M.A.; Paredes, L.C.; Peters, C.J. Treatment of bolivian hemorrhagic fever with intravenous ribavirin. Clin. Infect. Dis. 1997, 24, 718–722. [Google Scholar] [CrossRef]
- Weissenbacher, M.C.; Laguens, R.P.; Coto, C.E. Argentine hemorrhagic fever. Curr. Top. Microbiol. 1987, 134, 79–116. [Google Scholar]
- Kenyon, R.H.; Canonico, P.G.; Green, D.E.; Peters, C.J. Effect of ribavirin and tributylribavirin on argentine hemorrhagic fever (junin virus) in guinea pigs. Antimicrob. Agents Ch. 1986, 29, 521–523. [Google Scholar] [CrossRef]
- Salazar, M.; Yun, N.E.; Poussard, A.L.; Smith, J.N.; Smith, J.K.; Kolokoltsova, O.A.; Patterson, M.J.; Linde, J.; Paessler, S. Effect of ribavirin on junin virus infection in guinea pigs. Zoonoses Public Hlth. 2012, 59, 278–285. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grant, A.; Seregin, A.; Huang, C.; Kolokoltsova, O.; Brasier, A.; Peters, C.; Paessler, S. Junín Virus Pathogenesis and Virus Replication. Viruses 2012, 4, 2317-2339. https://doi.org/10.3390/v4102317
Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. Junín Virus Pathogenesis and Virus Replication. Viruses. 2012; 4(10):2317-2339. https://doi.org/10.3390/v4102317
Chicago/Turabian StyleGrant, Ashley, Alexey Seregin, Cheng Huang, Olga Kolokoltsova, Allan Brasier, Clarence Peters, and Slobodan Paessler. 2012. "Junín Virus Pathogenesis and Virus Replication" Viruses 4, no. 10: 2317-2339. https://doi.org/10.3390/v4102317
APA StyleGrant, A., Seregin, A., Huang, C., Kolokoltsova, O., Brasier, A., Peters, C., & Paessler, S. (2012). Junín Virus Pathogenesis and Virus Replication. Viruses, 4(10), 2317-2339. https://doi.org/10.3390/v4102317