Advanced Vaccine Candidates for Lassa Fever

Abstract
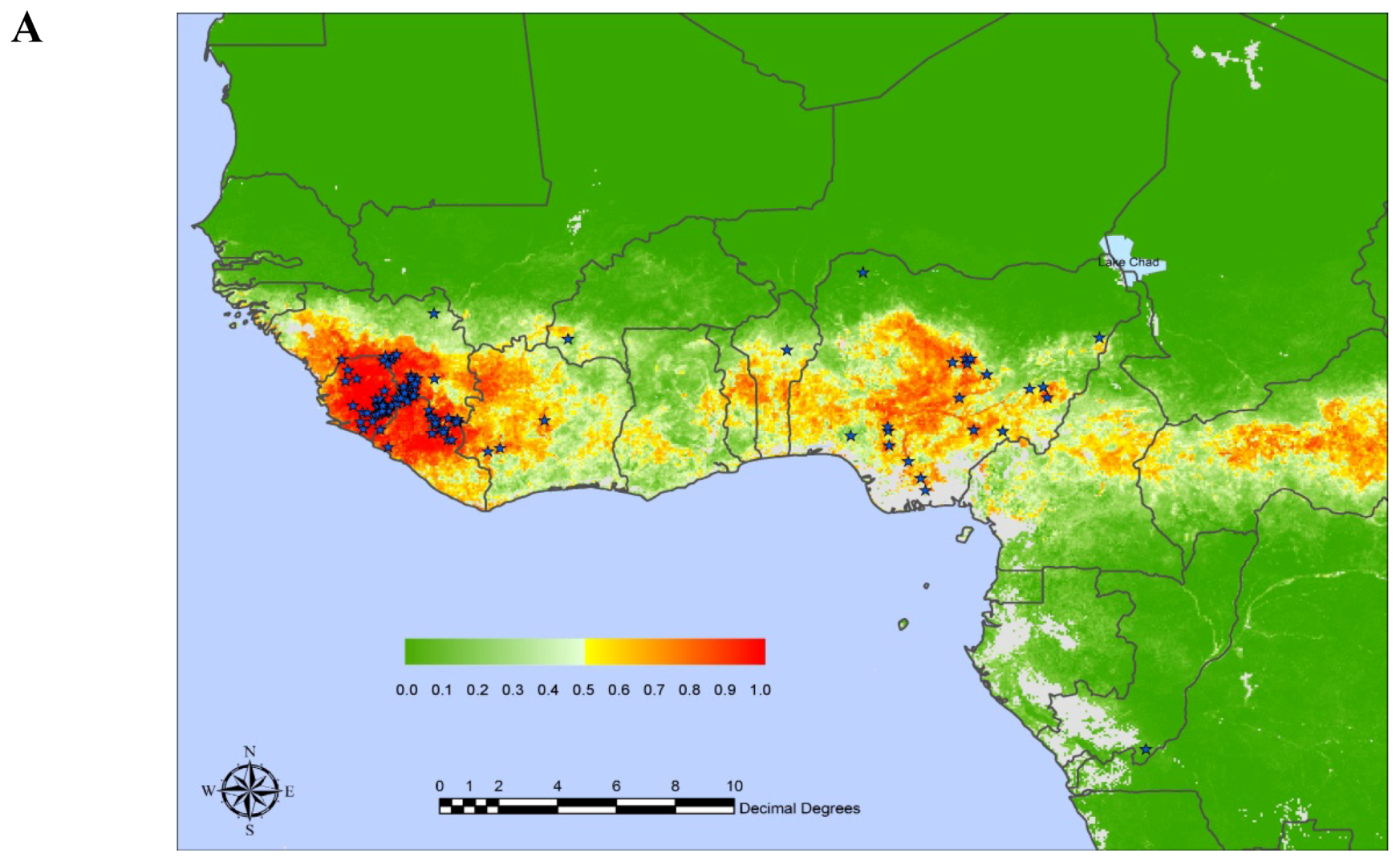
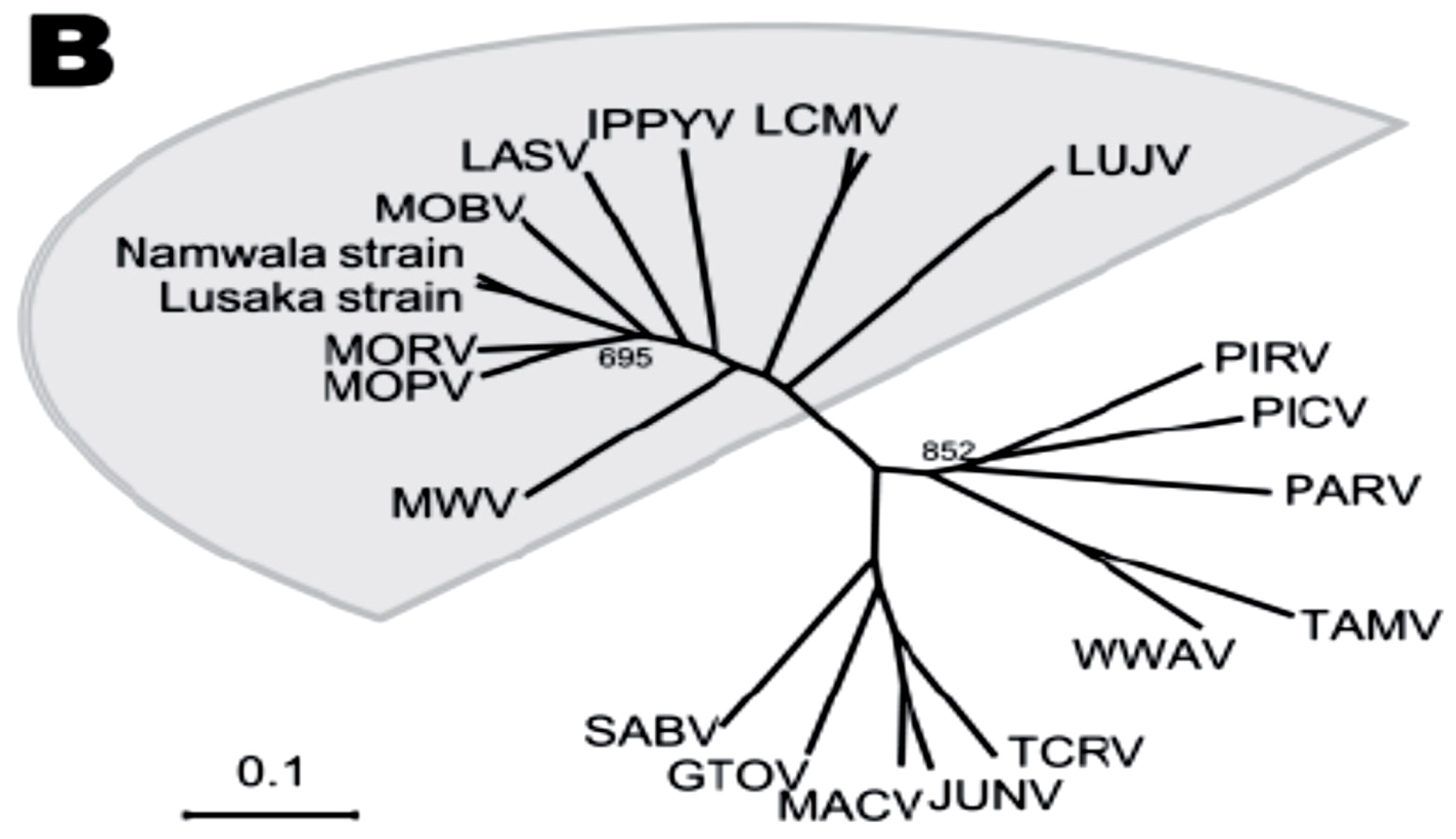
:1. Introduction


3. Lassa Fever Pathogenesis and Mechanisms of Protection
4. Non-Replication Competent Vaccine Platforms
4.1. “Killed” Vaccines and Virus-Like Particles
4.2. Peptide epitope-based vaccine approaches
4.3. Alphavirus Vector-Based Vaccines

4.4. DNA Immunization
4.5. Rationally Designed Replication-Competent Vaccines
4.5.1 Recombinant vaccinia virus expressing LASV antigens
4.5.2. Replication-competent VSV vectors expressing LASV glycoproteins
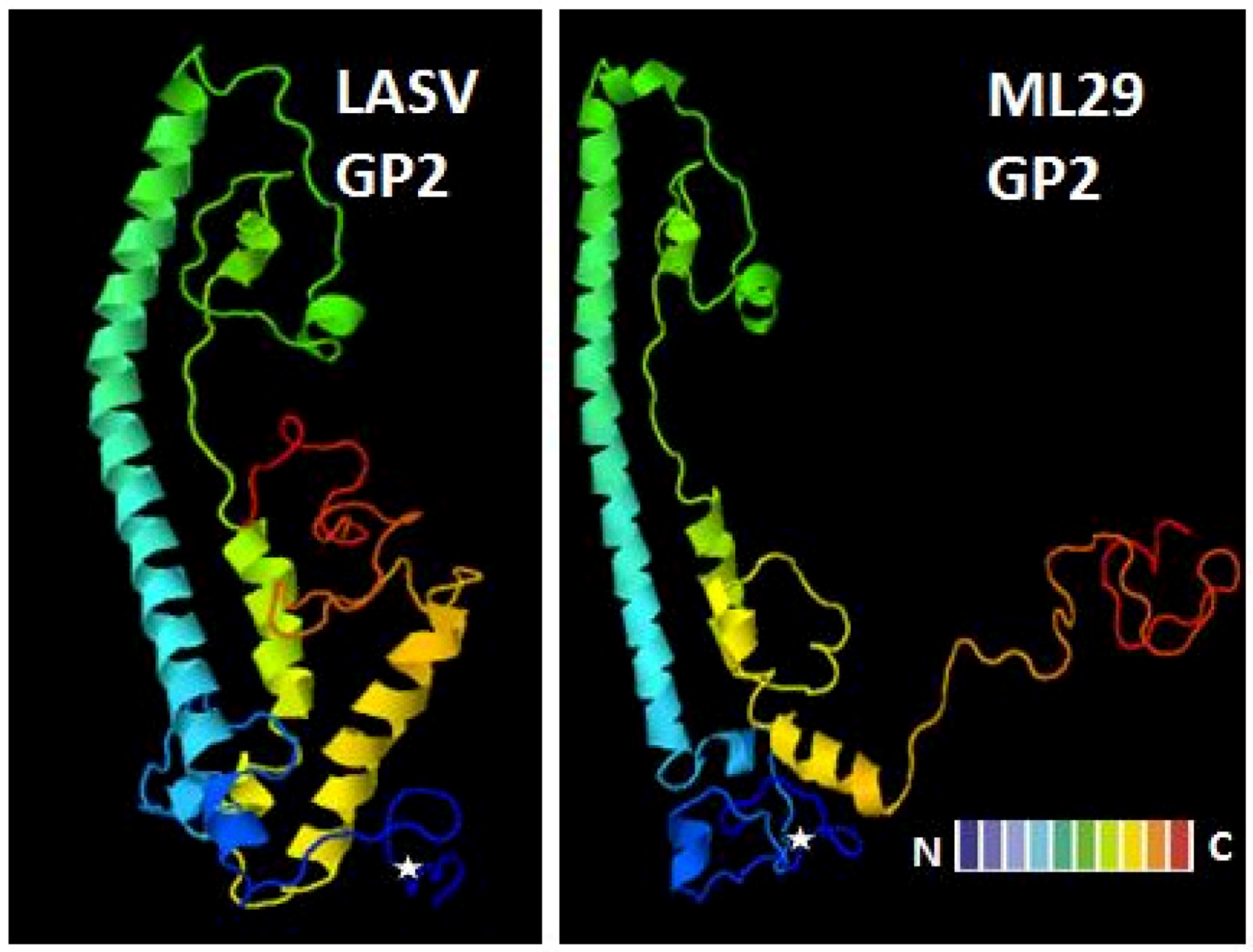
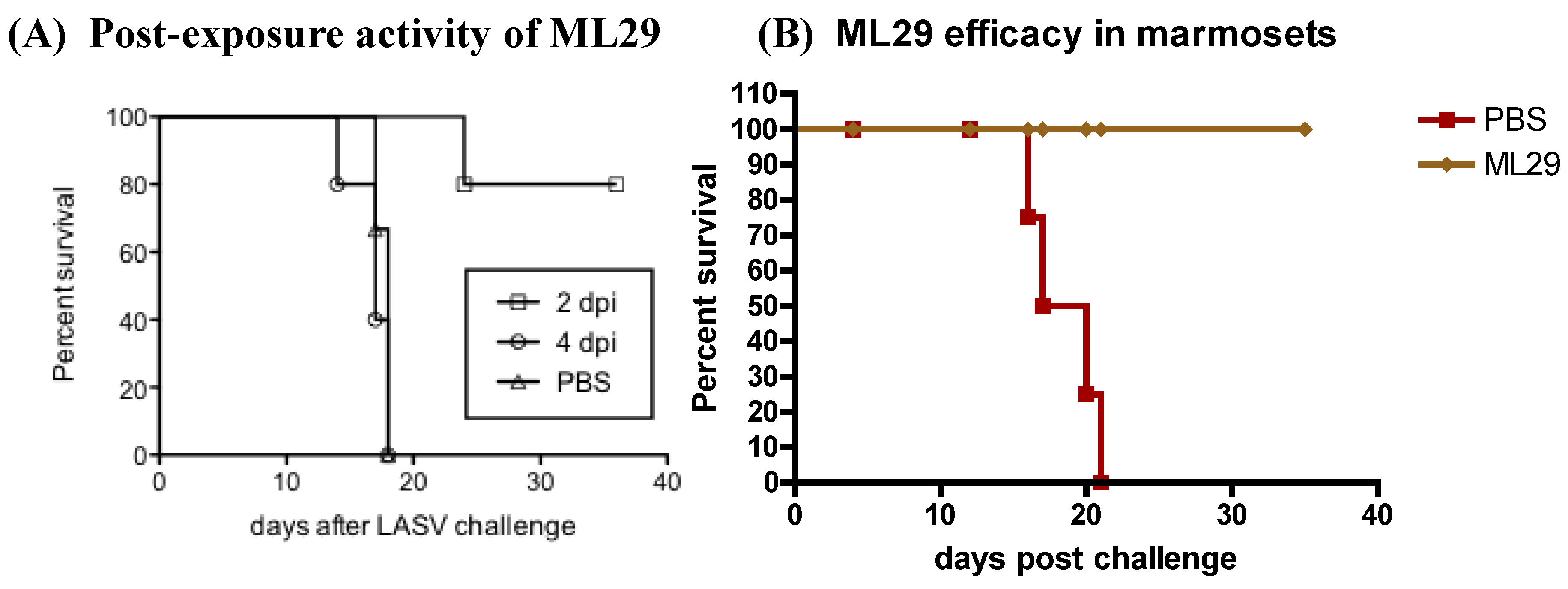
4.5.3. Reassortant vaccine platform, MOP/LAS (clone ML29)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations/Substitutions | Location | Putative function | Possible effects (References) |
|---|---|---|---|
| A6C, U8C | L RNA, 5’, 3’ NCR | Form panhandle | Enhance promoter stability [12] |
| A7264C | |||
| A7264C | |||
| G3328A | S RNA, 3’ NCR | Unknown | Deletions in the 3’NCR attenuate replication [158] |
| Y851N | L protein, N-term. | Cap-snatching | Attenuated transcription/ replication [162,163]? |
| N173S | NP, central domain | Interaction with RNA | Unknown |
| A485D | NP, C-terminal | Exonuclease | IFN modulation [164,165] |
| K272E | GP2, N-terminal | Fusion | Affect fusion/post-fusion events? |

| Animal Group | Challenged virus | Dose, PFU | Vacc/chal interval, days a | No. survived/No. infected | Survival, % | Day of death | |
|---|---|---|---|---|---|---|---|
| No vaccination | |||||||
| 1 | LASV-Jo | 10e+1 | na b | 0/4 | 0 | 15-17 | |
| 2 | LASV-Jo | 10e+3 | na | 0/5 | 0 | 15-16 | |
| 3 | LASV-803213 | 10e+3 | na | 0/5 | 0 | 13-15 | |
| 4 | LCMV-WE | 10e+3 | na | 0/5 | 0 | 13-14 | |
| The ML29 conventional vaccination (challenge on day 30) | |||||||
| 5 | 10e+2 | no challenge | na | na | 6/6 | 100 | na |
| 6 | 10e+6 | no challenge | na | na | 6/6 | 100 | na |
| 7 | 10e+3 | LASV-Jo | 10e+3 | 30 | 6/6 | 100 | na |
| 8 | 10e+3 | LASV-803213 | 10e+3 | 30 | 5/5 | 100 | na |
| 9 | 10e+3 | LCMV-WE | 10e+3 | 30 | 0/6 | 0 | 16-21 |
| The simultaneous vaccination/challenge experiments (challenge on day 0 and 2) | |||||||
| 10a. | 10e+6 | LASV-Jo | 10e+1 | 0 | 5/5 | 100 | na |
| 10b. | 10e+6 | LASV-Jo | 10e+1 | 2 | 3/5 | 60 | 10c,15 |
| 11a. | 10e+6 | LASV-Jo | 10e+3 | 0 | 4/4 | 100 | na |
| 11b. | 10e+6 | LASV-Jo | 10e+3 | 2 | 4/5 | 80 | 10 c |
| 12a. | 10e+2 | LASV-Jo | 10e+3 | 0 | 3/4 | 75 | 14 |
| 12b. | 10e+2 | LASV-Jo | 10e+3 | 2 | 3/4 | 75 | 16 |
| 13 | 10e+6 | LASV-803213 | 10e+3 | 0 | 3/5 | 60 | 12,17 |
| 14 | 10e+6 | LCMV-WE | 10e+3 | 0 | 0/5 | 0 | 14-16 |

| Animal No | Time, days after challenge: | |||||
|---|---|---|---|---|---|---|
| 0 | 5 | 15 | 17-19 | 20 | 35 | |
| Non-vaccinated | ||||||
| CJ25391 | <1.0 | 2.95 | 4.32 | nd | na | na |
| CJ26255 | <1.0 | + | 4.45 | 5.54 | na | na |
| CJ26494 | <1.0 | 2.11 | 5.38 | nd | na | na |
| CJ26950 | <1.0 | + | 4.81 | 6.78 | na | na |
| ML29-vaccinated | ||||||
| CJ26493 | <1.0 | 2.67 | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ27008 | <1.0 | <1.0/- | <1.0/ + | <1.0/- | <1.0/- | <1.0/- |
| CJ26190 | <1.0 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26946 | <1.0 | <1.0/+ | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26256 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26249 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
4.5.4. Recombinant Yellow Fever 17D expressing LASV antigens.
5. Current challenges and problems to overcome
5.1. The FDA Animal Rule
5.2. Vaccine formulation and target population
5.3. Conclusions and prospects for future
Acknowledgments
Conflict of Interest
References
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immuno.l 2002, 262, 75–109. [Google Scholar]
- Khan, S.H.; Goba, A.; Chu, M. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antivir. Res. 2008, 78, 103–115. [Google Scholar]
- Fichet-Calvet, E.; Rogers, D.J. Risk Maps of Lassa Fever in West Africa. PLoS Negl. Trop. Dis. 2009, 3. [Google Scholar]
- Loureiro, M.E.; Wilda, M.; Levingston-Macleod, J.M. Molecular determinants of Arenavirus Z protein homo-oligomerization and L polymerase binding. J. Virol. 2011. JVI.05691-05611.. [Google Scholar]
- Richmond, J.K.; Baglole, D. Lassa fever: epidemiology, clinical features, and social consequences. BMJ 2003, 327, 1271–1275. [Google Scholar] [CrossRef]
- Safronetz, D.; Job, E.; Nafomon, S.; Traore, S.F.; Raffel, S.J.; Fischer, E.R.; Ebihara, H.; Branco, L.; Garry, R.F.; Schwan, T.G.; Feldmann, H. Detection of Lassa Virus. Mali. Emerg. Infect. Dis. 2010, 16, 1123–1126. [Google Scholar] [CrossRef]
- Andersen, K.G.; Shylakhter, I.; Tabrizi, S.; Grossman, S.R.; Happi, C.T.; Sabeti, P.C. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos. T. R. Soc. B. 2012, 367, 868–877. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine: A review. Expert. Rev. Vaccines 2004, 3, 103–111. [Google Scholar]
- Ishii, A.; Thomas, Y.; Moonga, L.; Nakamura, I.; Ohnuma, A.; Hang'ombe, B.; Takada, A.; Mweene, A.; Sawa, H. Novel arenavirus, Zambia. Emerg. Infect. Dis. 2011, 17, 1921–1924. [Google Scholar] [CrossRef]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J. Arenaviridae. In Virus Taxonomy, VIIIth Report of the ICTV; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2005; pp. 725–733. [Google Scholar]
- Lukashevich, I.S.; Salvato, M.S. Lassa Virus Genome. Current Genomics 2006, 7, 351–379. [Google Scholar]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J. Arenaviridae. In Virus Taxonomy, VIIIth Report of the ICTV; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2005; pp. 725–733. [Google Scholar]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J.; Charrel, R.N.; Gonzalez, J.P.; Lukashevich, I.S.; Peters, C.J.; Romanowski, V. Family Arenaviridae, Virus Taxonomy, Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; Andrew King, M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press, Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 715–723. [Google Scholar]
- Charrel, R.N.; de Lamballerie, X.; Emonet, S. Phylogeny of the genus Arenavirus. Curr. Opin. Microbiol. 2008, 11, 362–368. [Google Scholar]
- Emonet, S.F.; de la Torre, J.C.; Domingo, E.; Sevilla, N. Arenavirus genetic diversity and its biological implications. Infect. Genet. Evol. 2009, 9, 417–429. [Google Scholar] [CrossRef]
- Charrel, R.N.; Lemasson, J.J.; Garbutt, M. New insights into the evolutionary relationships between arenaviruses provided by comparative analysis of small and large segment sequences. Virology 2003, 317, 191–196. [Google Scholar]
- Bowen, M.; Rollin, P.; Ksiazek, T. Genetic diversity among Lassa virus strains. J. Virol. 2000, 74, 6992–7004. [Google Scholar] [CrossRef]
- Lozano, M.E.; Posik, D.M.; Albarino, C.G. Characterization of arenaviruses using a family-specific primer set for RT-PCR amplification and RFLP analysis. Its potential use for detection of uncharacterized arenaviruses. Virus Res. 1997, 49, 79–89. [Google Scholar] [CrossRef]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. Phylogenetic analysis of the Arenaviridae: patterns of virus evolution and evidence for cospeciation between arenaviruses and their rodent hosts. Mol. Phylogenet. Evol. 1997, 8, 301–316. [Google Scholar]
- Gunther, S.; Emmerich, P.; Laue, T. Imported Lassa fever in Germany: molecular characterization of a new Lassa virus strain. Emerg. Infect. Dis. 2000, 6, 466–476. [Google Scholar] [CrossRef]
- Vieth, S.; Torda, A.E.; Asper, M.; Schmitz, H.; Gunther, S. Sequence analysis of L RNA of Lassa virus. Virology 2004, 318, 153–168. [Google Scholar] [CrossRef]
- Emonet, S.; Lemasson, J.J.; Gonzalez, J.P.; Lamballerie, X.d.; Charrel, R.N. Phylogeny and evolution of old world arenaviruses. Virology 2006, 350, 251–257. [Google Scholar]
- Clegg, J. Molecular phylogeny of the Arenaviridae. Curr. Top. Microbiol. Immunol. 2002, 262, 1–24. [Google Scholar] [CrossRef]
- Lalis, A.; Leblois, R.; Lecompte, E.; Denys, C.; ter Meulen, J.; Wirth, T. The Impact of Human Conflict on the Genetics of Mastomys natalensis and Lassa Virus in West Africa. PLoS One 2012, 7, e37068. [Google Scholar]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Peters, C.J.; Jahrling, P.B.; Liu, C.T.; Kenyon, R.H.; McKee, K.T.; Oro, J.G.B. Experimental studies of arenaviral hemorrhagic fevers. Curr. Top. Microbiol. Immunol. 1987, 134, 5–68. [Google Scholar]
- Swanepoel, R.; Leman, P.A.; Shepherd, A.J.; Shepherd, S.P.; Kiley, M.P.; McCormick, J.B. Identification of Ippy as a Lassa-fever-related virus. Lancet 1985, 1, 639. [Google Scholar]
- Meunier, D.Y.; McCormick, J.B.; Georges, A.J.; Georges, M.C.; Gonzalez, J.P. Comparison of Lassa, Mobala, and Ippy virus reactions by immunofluorescence test. Lancet. 1985, 1, 873–874. [Google Scholar]
- Gonzalez, J.; McCormick, J.B.; Saluzzo, J.F.; Herve, J.P.; Georges, A.J.; Johnson, K.M. An arenavirus isolated from wild-caught rodents (Pramys species) in the Central African Republic. Intervirol. 1983, 19, 105–112. [Google Scholar] [CrossRef]
- Wulff, H.B.M.; McIntosh, D.B.; Hamner & Johnson, K.M. Isolation of an arenavirus closely related to lassa virus from rodents. Bull. WHO. 1977, 55, 441–444. [Google Scholar]
- Johnson, K.M.; Taylor, P.; Elliott, L.H.; Tomori, O. Recovery of a Lassa-Related Arenavirus in Zimbabwe. Am. J. Trop. Med. Hyg. 1981, 30, 1291–1293. [Google Scholar]
- Georges, A.; Gonzalez, J.P.; Abdul-Wahid, S.; Saluzzo, J.F.; Meunier, D.M.; McCormick, J.B. Antibodies to Lassa and Lassa-like viruses in man and mammals in the Central African Republic. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 78–79. [Google Scholar] [CrossRef]
- Kiley, M.P.; Lange, J.V.; Johnson, K.M. Protection of rhesus monkeys from Lassa virus by immunisation with closely related Arenavirus. Lancet 1979, 2, 738. [Google Scholar]
- Walker, D.H.; Johnson, K.M.; Lange, J.V.; Gardner, J.J.; Kiley, M.P.; McCormick, J.B. Experimental infection of rhesus monkeys with Lassa virus and a closely related arenavirus, Mozambique virus. J. Infect. Dis. 1982, 146, 360–363. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine. Expert Rev. Vaccines 2004, 3, 103–111. [Google Scholar]
- Salazar-Bravo, J.; Ruedas, L.A.; Yates, T.L. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 2002, 262, 25–63. [Google Scholar]
- Keenlyside, R.A.; McCormick, J.B.; Webb, P.A.; Smith, E.; Elliott, L.; Johnson, K.M. Case-control study of Mastomys natalensis and humans in Lassa virus-infected households in Sierra Leone. Am. J.Trop. Med. Hyg. 1983, 32, 829–837. [Google Scholar]
- Demby, A.H.; Inapogui, A.; Kargbo, K.; Koninga, J.; Kourouma, K.; Kanu, J.; Coulibaly, M.; Wagoner, K.D.; Ksiazek, T.G.; Peters, C.J.; Rollin, P.E.; Bausch, D.G. Lassa fever in Guinea: II. Distribution and prevalence of Lassa virus infection in small mammals. Vector Borne Zoonotic Dis. 2001, 1, 283–297. [Google Scholar] [CrossRef]
- Demartini, J.; Green, D.E.; Monath, T.P. Lassa virus infection of Mastomy natalensis in Sierra Leone. Gros and microscopical findings in infected and uninfected animals. Bull World Health Organ 1975, 52, 651–663. [Google Scholar]
- Lecompte, E.; Meulen, J.T.; Emonet, S.; Daffis, S.; Charrel, R.N. Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology 2007, 364, 178–183. [Google Scholar] [CrossRef]
- Paweska, J.; Sewlall, N.H.; Ksiazek, T.G.; Blumberg, L.H.; Hale, M.J.; Lipkin, W.I. Nosocomial outbreak of novel arenavirus infection, southern Africa. Emerg. Infect. Dis. 2009, 15, 1598–1602. [Google Scholar]
- Briese, T.; Paweska, J.T.; McMullan, L.K. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever-Associated Arenavirus from Southern Africa. PLoS. Pathog. 2009, 4, e1000455. [Google Scholar]
- Palacios, G.; Savji, N.; Hui, J. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J. Gen. Virol. 2010, 91, 1315–1324. [Google Scholar] [CrossRef]
- Coulibaly-N'Golo, D.; Allali, B.; Kouassi, S.K. Novel Arenavirus Sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Co´te d'Ivoire: Implications for Evolution of Arenaviruses in Africa. PLoS ONE 2011, 6, e20893. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Sanders, C.; Kistler, A.L. Identification, Characterization, and In Vitro Culture of Highly Divergent Arenaviruses from Boa Constrictors and Annulated Tree Boas: Candidate Etiological Agents for Snake Inclusion Body Disease. mBio 2012, 3. [Google Scholar]
- Kerber, R.; Rieger, T.; Busch, C. Cross-species analysis of the replication complex of Old World arenaviruses reveals two sites in nucleoprotein involved in L protein function. J. Virol. 2011, 85, 12518–12528. [Google Scholar]
- Lukashevich, I.S. Generation of reassortants between African arenaviruses. Virology 1992, 188, 600–605. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; A.D. Vasiuchkov, T.A.; Stel'makh, E.P.; Scheslenok & Shabanov, A.G. The isolation and characteristics of reassortants between the Lassa and Mopeia arenaviruses. Vopr. Virusol. 1991, 36, 146–150. [Google Scholar]
- Cummins, D.; McCormick, J.; Bennett, D. Acute sensorineural deafness in Lassa fever. JAMA. 1990, 264, 2093–2096. [Google Scholar] [CrossRef]
- Kitching, A.; Addiman, S.; Cathcart, S. A fatal case of Lassa Fever in London, January 2009. EUROSURVEI L LANCE 2009, 14, 1–3. [Google Scholar]
- Moraz, M.-L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Review of Anti-infective Therapy 2011, 9, 49–59. [Google Scholar] [CrossRef]
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 2006, 8, 1194–1202. [Google Scholar]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J. Virol. 2009. JVI.01948-01908.. [Google Scholar]
- Hayes, M.W.; Carrion, R.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World Arenaviruses Inhibit TLR2/Mal-Dependent Pro-Inflammatory Cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Maryankova, R.F.; Vladyko, A.S. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-a gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; Mcrae, M.; Rollin, P.E.; Pulendran, B. Cutting Edge: Impairment of Dendritic Cells and Adaptive Immunity by Ebola and Lassa Viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar]
- Meulen, J.; Badusche, M.; Satoguina, J. Old and New World arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by Lassa virus-specific human CD4+ T-cell clones. Virology 2004, 321, 134–143. [Google Scholar]
- Ter Meulen, J.; Badusche, M.; Kuhnt, K. Characterization of human CD4+ T-cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar]
- Jahrling, P.B.; Frame, J.D.; Smith, S.B.; Monson, M.H. Endemic Lassa fever in Liberia. III. Characterization of Lassa virus isolates. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 374–379. [Google Scholar] [CrossRef]
- Walker, D.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Knobloch, J.M.J.; Webb, P.A.; Dietrich, M.; Schumacher, H.H.; Dennis, E. Clinical observations in 42 patients with Lassa fever. Tropenmed Parasitol. 1980, 31, 389–398. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Elliott, L.H.; Whitfield, S.G.; Johnson, K.M. Lassa virus hepatitis: a study of fatal Lassa fever in humans. Am. J. Trop. Med. Hyg. 1986, 35, 401–407. [Google Scholar]
- Geisberg, T.W.; Jahrling, P.B. Exotic emerging viral diseases: progress and challenges. Nature Medicine 2004, 10, 110–S121. [Google Scholar]
- Ter Meulen, J.; Lukashevich, I.S.; Sidibe, K. Hunting of peridomestic rodents and consumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am. J. Trop. Med. Hyg. 1996, 55, 661–666. [Google Scholar]
- Dylla, D.E.; Michele, D.E.; Campbell, K.P.; McCray, P.B., Jr. Basolateral Entry and Release of New and Old World Arenaviruses from Human Airway Epithelia. J. Virol. 2008, 82, 6034–6038. [Google Scholar]
- Schlie, K.; Maisa, A.; Freiberg, F.; Groseth, A.; Strecker, T.; Garten, W. Viral Protein Determinants of Lassa Virus Entry and Release from Polarized Epithelial Cells. J. Virol. 2010, 84, 3178–3188. [Google Scholar] [CrossRef]
- Kunz, S. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology 2009, 387, 245–249. [Google Scholar] [CrossRef]
- Cosset, F.-L.; Marianneau, P.; Verney, G. Characterization of Lassa Virus Cell Entry and Neutralization with Lassa Virus Pseudoparticles. J. Virol. 2009, 83, 3228–3237. [Google Scholar]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef]
- McCormick, J.B. Lassa fever. In Factors in Emergence and Control of Rodent-borne Viral Diseases (Hantaviral and Arenal Diseases) 1999; Saluzzo, J.F., Dodet, B., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 177–195. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P. Arenavirus pathophysiology. In The Arenaviridae; Salvato, M.S., Ed.; Plenum Press, 1993; pp. 299–323. [Google Scholar]
- Callis, R.T.; Jahrling & DePaoli, P.B. A Pathology of Lassa virus infection in the rhesus monkey. Am. J. Trop. Med. Hyg. 1982, 31, 1038–1045. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Eddy, G.A.; Johnson, K.M.; Callis, R.T.; Stephen, E.L. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 1980, 141, 580–589. [Google Scholar] [CrossRef]
- Lange, J.V.; Mitchell, S.W.; McCormick, J.B.; Walker, D.H.; Evatt, B.L.; Ramsey, R.R. Kinetic study of platelets and fibrinogen in Lassa virus-infected monkeys and early pathologic events in Mopeia virus-infected monkeys. Am. J. Trop. Med. Hyg. 1985, 34, 999–1007. [Google Scholar]
- Kunz, S. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb. Haemost 2009, 102, 1024–1029. [Google Scholar]
- Lukashevich, I.S.; I. Tikhonov, I.; Rodas, J.D. Arenavirus-mediated liver pathology: acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, I.I. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P. Early and Strong Immune Responses Are Associated with Control of Viral Replication and Recovery in Lassa Virus-Infected Cynomolgus Monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar]
- Pannetier, D.; Reynard, S.; Russier, M. Human Dendritic Cells Infected with the Nonpathogenic Mopeia Virus Induce Stronger T-Cell Responses than Those Infected with Lassa Virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef]
- Carrion, R.; Brasky, K.; Mansfield, K. Lassa Virus Infection in Experimentally Infected Marmosets: Liver Pathology and Immunophenotypic Alterations in Target Tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef]
- Zapata, J.C.; Pauza, C.D.; Djavani, M.M. Lymphocytic choriomeningitis virus (LCMV) infection of macaques: A model for Lassa fever. Antivir. Res. 2011, 92, 125–138. [Google Scholar]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar]
- Zhou, S.; Kurt-Jones, E.A.; Cerny, A.M.; Chan, M.; Bronson, R.T.; Finberg, R.W. MyD88 Intrinsically Regulates CD4 T-Cell Responses. J. Virol. 2009, 83, 1625–1634. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B.A. Identification of -dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Kunz, S.; Rojek, J.M.; Kanagawa, M. Posttranslational Modification of {alpha}-Dystroglycan, the Cellular Receptor for Arenaviruses, by the Glycosyltransferase LARGE Is Critical for Virus Bindin. J. Virol. 2005, 79, 14282–14296. [Google Scholar] [CrossRef]
- Oldstone, M.B.A.; Campbell, K.P. Decoding arenavirus pathogenesis: Essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology 2011, 411, 170–179. [Google Scholar] [CrossRef]
- Imperiali, M.; Spörri, R.; Hewitt, J.; Oxenius, A. Post-translational modification of α-dystroglycan is not critical for lymphocytic choriomeningitis virus receptor function in vivo. J. Gen. Virol. 2008, 89, 2713–2722. [Google Scholar]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J Virol. 2012, 86, 2067–2078. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell surface molecules involved in infection mediated by lymphocytic choriomeningitis virus glycoprotein. J. Vet. Med. Sci. 2012. [Google Scholar]
- Clegg, J.C.; Lloyd, G. Vaccinia recombinant expressing Lassa-virus internal nucleocapsid protein protects guinea pigs against Lassa fever. Lancet. 1987, 2, 186–188. [Google Scholar] [CrossRef]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliot, L.H.; King, I.J. Clinical virology of Lassa fever in hospitalized patient. J. Infect. Dis. 1987, 155, 456–463. [Google Scholar]
- Pushko, P.; Geisbert, J.; Parker, M.; Jahrling, P.; Smith, J. Individual and bivalent vaccines based on alphavirus replicons protect guinea pigs against infection with Lassa and Ebola viruses. J. Virol. 2001, 75, 11677–11685. [Google Scholar]
- McCormick, J.; Mitchell, S.; Kiley, M.; Ruo, S.; Fisher-Hoch, S. Inactivated Lassa virus elicits a non protective immune response in rhesus monkeys. J. Med. Virol. 1992, 31, 1–7. [Google Scholar]
- Lukashevich, I.S.; Clegg, J.C.; Sidibe, K. Lassa virus activity in Guinea: distribution of human antiviral antibody defined using enzyme-linked immunosorbent assay with recombinant antigen. J. Med. Virol. 1993, 40, 210–217. [Google Scholar] [CrossRef]
- Bausch, D.G.; Rollin, P.E.; Demby, A.H. Diagnosis and Clinical Virology of Lassa Fever as Evaluated by Enzyme-Linked Immunosorbent Assay, Indirect Fluorescent-Antibody Test, and Virus Isolation. J. Clin. Microbiol. 2000, 38, 2670–2677. [Google Scholar]
- Branco, L.M.; Grove, J.N.; Boisen, M.L.; Shaffer, J.G.; Goba, A.; Fullah, M.; Momoh, M.; Grant, D.S.; Garry, R.F. Emerging trends in Lassa fever: redefining the role of immunoglobulin M and inflammation in diagnosing acute infection. Virol. J. 2011, 8. [Google Scholar]
- Krasnianskiĭ, V.; Potryvaeva, N.V.; Borisevich, I.V.; Gradoboev, V.N.; Pashanina, T.P.; Pshenichnov, V.A. A trial to produce an inactivated Lassa fever vaccine. Vopr Virusol. 1993, 38(6), 276–279. [Google Scholar]
- Branco, L.; Grove, J.; Geske, F. Lassa virus-like particles displaying all major immunological determinants as a vaccine candidate for Lassa hemorrhagic fever. Virology J. 2010, 7, 279. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. Rev. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef]
- Borrow, P.; Martinez-Sobrido, L.; De la Torre, J.C. Inhibition of the Type I Interferon Antiviral Response During Arenavirus Infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef]
- Koyama, S.; Coban, C.; Aoshi, T.; Horii, T.; Akira, S.; Ishii, K.J. Innate immune control of nucleic acid-based vaccine immunogenicity. Expert. Rev. Vaccines. 2009, 8, 1099–10107. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E. Junín Virus Infection Activates the Type I Interferon Pathway in a RIG-I-Dependent Manner. PLoS Negl Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingström, J. Processing of Genome 5′ Termini as a Strategy of Negative-Strand RNA Viruses to Avoid RIG-I-Dependent Interferon Induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef]
- Walsh, K.; Teijaro, J.R.; Zuniga, E.I.; Welch, M.J.; Fremgen, D.M.; Blackburn, S.D.; von Tiehl, K.F.; Wherry, E.J.; Flavell, R.A.; Oldstone, M.B. Toll-like Receptor 7 Is Required for Effective Adaptive Immune Responses that Prevent Persistent Virus Infection. Cell Host Microbe. 2012, 11, 643–653. [Google Scholar] [CrossRef]
- Macal, M.; Lewis, G.M.; Kunz, S.; Flavell, R.; Harker, J.A.; Zúñiga, E.I. Plasmacytoid Dendritic Cells Are Productively Infected and Activated through TLR-7 Early after Arenavirus Infection. Cell Host Microbe. 2012, 11, 617–630. [Google Scholar] [CrossRef]
- Baz Morelli, A.; Becher, D.; Koernig, S.; Silva, A.; Drane, D.; Maraskovsky, E. ISCOMATRIX: a novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. Journal of Medical Microbiology 2012, 61, 935–943. [Google Scholar] [CrossRef]
- Boesen, A.; Sundar, K.; Coico, R. Lassa Fever Virus Peptides Predicted by Computational Analysis Induce Epitope-Specific Cytotoxic-T-Lymphocyte Responses in HLA-A2.1 Transgenic Mice. Clin. Diagn. Lab. Immunol. 2005, 12, 1223–1230. [Google Scholar]
- Botten, J.; Alexander, J.; Pasquetto, V. Identification of Protective Lassa Virus Epitopes That Are Restricted by HLA-A2. J. Virol. 2006, 80, 8351–8361. [Google Scholar] [CrossRef]
- Botten, J.; Whitton, J.L.; Barrowman, P. A Multivalent Vaccination Strategy for the Prevention of Old World Arenavirus Infection in Humans. J. Virol. 2010, 84, 9947–9956. [Google Scholar] [CrossRef]
- Liu, F.; Feuer, R.; Hassett, D.E.; Whitton, J.L. Peptide vaccination of mice immune to LCMV or vaccinia virus causes serious CD8+ T cell-mediated, TNF-dependent immunopathology. J. Clin. Invest. 2006, 116, 465–475. [Google Scholar]
- Strauss, J.; Strauss, E. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar]
- Vander Veen, R.L.; Hank Harris, D.L.; Kurt, I. Kamrud. Alphavirus replicon vaccines. Animal Health Res. Rev. 2012. [Google Scholar]
- Bergen, M.J.; Pan, C.-H.; Greer, C.E.; Legg, H.S.; Polo, J.M.; Griffin, D.E. Comparison of the Immune Responses Induced by Chimeric Alphavirus-Vectored and Formalin-Inactivated Alum-Precipitated Measles Vaccines in Mice. PLoS ONE 2010, 5, e10297. [Google Scholar]
- Carroll, T.; Matzinger, S.R.; Barro, M.; Fritts, L.; McChesney, M.B.; Miller, C.J.; Johnston, R.E. Alphavirus replicon-based adjuvants enhance the immunogenicity and effectiveness of Fluzone® in rhesus macaques. Vaccine 2011, 29, 931–940. [Google Scholar]
- Thompson, J.; Whitmore, A.C.; Konopka, J.L.; Collier, M.L.; Richmond, E.M.; Davis, N.L. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. U. S. A. 2006, 7, 3722–3727. [Google Scholar]
- Thompson, J.; Nicholson, M.G.; Whitmore, A.C.; Zamora, M.; West, A.; Iwasaki, A. Nonmucosal alphavirus vaccination stimulates a mucosal inductive environment in the peripheral draining lymph node. J. Immunol. 2008, 181, 574–585. [Google Scholar]
- Thompson, J.M.; Whitmore, A.C.; Staats, H.F.; Johnston, R.E. Alphavirus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine 2008, 26, 4267–4275. [Google Scholar] [CrossRef]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 2, 389–401. [Google Scholar]
- Nelson, E.; Prieto, D.; Alexander, T.G.; Pushko, P. Venezuelan equine encephalitis replicon immunization overcomes intrinsic tolerance and elicits effective anti-tumor immunity to the 'self' tumor-associated antigen, neu in a rat mammary tumor model. Breast Cancer Res. Treat. 2003, 82, 169–183. [Google Scholar] [CrossRef]
- Johnston, R.; Davis, N.; Smith, J.; Pushko, P.; Parker, M.; Ludwig, G. Alphavirus RNA replicon systems. U.S. Patent 7,235,235, 2007. Alphavax IP. [Google Scholar]
- Pushko, P. Vector platforms derived from the alphavirus vaccines. U.S. Patent Application No. 2006/0198854 2006. [Google Scholar]
- Rodriguez-Carreno, M.P.; Nelson, M.S.; Botten, J.; Smith-Nixon, K.; Buchmeier, M.J.; Whitton, J.L. Evaluating the immunogenicity and protective efficacy of a DNA vaccine encoding Lassa virus nucleoprotein. Virology 2005, 335, 87–98. [Google Scholar] [CrossRef]
- Grant-Klein, R.; Altamura, L.A.; Schmaljohn, C.S. Progress in recombinant DNA-derived vaccines for Lassa virus and filoviruses. Virus Res. 2011, 162, 148–161. [Google Scholar]
- Whitton, J.L.; Tishon, A.; Lewicki, H. Molecular analyses of a five-amino-acid cytotoxic T-lymphocyte (CTL) epitope: an immunodominant region which induces nonreciprocal CTL cross-reactivity. J. Virol. 1989, 63, 4303–4310. [Google Scholar]
- Schulz, M.; Aichele, P.; Schneider, R.; Hansen, T.H.; Zinkernagel, R.M.; Hengartner, H. Major histocompatibility complex binding and T cell recognition of a viral nonapeptide containing a minimal tetrapeptide. Eur. J. Immunol. 1991, 21, 1181–1185. [Google Scholar] [CrossRef]
- Djavani, M.; Yin, C.; Xia, L.; Lukashevich, I.S.; Pauza, C.; Salvato, M. Murine immune responses to mucosally delivered Salmonella expressing Lassa fever virus nucleoprotein. Vaccine. 2000, 18, 1543–1554. [Google Scholar] [CrossRef]
- Djavani, M.; Yin, C.; Lukashevich, I.; Rodas, J.; Rai, S.; Salvato, M. Mucosal immunization with Salmonella typhimurium expressing Lassa virus nucleocapsid protein cross-protects mice from lethal challenge with lymphocytic choriomeningitis virus. J. Hum. Virol. 2001, 4, 103–108. [Google Scholar]
- Whitton, J.L.; Sheng, N.; Oldstone, M.B.; McKee, T.A. A "string-of-beads" vaccine, comprising linked minigenes, confers protection from lethal-dose virus challenge. Journal of Virology 1993, 67, 348–352. [Google Scholar]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert. Rev. Vaccines 2012, 11, 189–2012. [Google Scholar]
- Lauring, A.S.; Jeremy, O. Raul Andino. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Lukashevich, I.S.; Patterson, J.; Carrion, R. A live attenuated vaccine for Lassa fever made by reassortment of Lassa and Mopeia viruses. J. Virol. 2005, 79, 13934–13942. [Google Scholar] [CrossRef]
- Ambrosio, A.; Saavedra, M.C.; Mariani, M.A.; Gamboa, G.S.; Maiza, A.S. Argentine hemorrhagic fever vaccines. Human Vaccines 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Auperin, D.D.; Esposito, A.A.; Lange, J.V. Construction of a recombinant vaccinia virus expressing the Lassa virus glycoprotein gene and protection of guinea pigs from a lethal Lassa virus infection. Virus Res. 1988, 9, 233–248. [Google Scholar]
- Fisher-Hoch, S.P.; Hutwagner, L.; Brown, B.; McCormick, J.B. Effective Vaccine for Lassa Fever. J. Virol. 2000, 74, 6777–6783. [Google Scholar]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Möller, P.; Wagner, R.; Volchkov, V.; Klenk, H.D.; Feldmann, H.; Ströher, U. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jones, S.; Fritz, E.A. Development of a new vaccine for the prevention of Lassa Fever. PLoS Medicine 2005, 2, e183. [Google Scholar] [CrossRef] [Green Version]
- Lukashevich, I.S.; Patterson, J. Safety, immunogenicity, and efficacy of the ML29 vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar]
- Bredenbeek, P.J.; Molenkamp, R.; Spaan, W.J.M. A recombinant Yellow Fever 17D vaccine expressing Lassa virus glycoproteins. Virology 2006, 345, 299–304. [Google Scholar] [CrossRef]
- Jiang, X.; Dalebout, T.J.; Bredenbeek, P.J. Yellow fever 17D-vectored vaccines expressing Lassa virus GP1 and GP2 glycoproteins provide protection against fatal disease in guinea pigs. Vaccine 2011, 29, 1248–1257. [Google Scholar] [CrossRef]
- Morrison, H.G.; Bauer, S.P.; Lange, J.V.; Esposito, J.J.; McCormick, J.B.; Auperin, D.D. Protection of guinea pigs from Lassa fever by vaccinia virus recombinants expressing the nucleoprotein or the envelope glycoproteins of Lassa virus. Virology 1989, 171, 179–188. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B.; Auperin, D. Protection of rhesus monkeys from fatal Lassa fever by vaccination with a recombinant vaccinia virus containing the Lassa virus glycoprotein gene. Proc. Natl. Acad. Sci. USA 1989, 86, 317–321. [Google Scholar]
- Tani, H.; Shigeru, M.; Yoshiharu, M. Development and applications of VSV vectors based on cell tropism. Front Microbiol. 2011. [Google Scholar]
- Geisbert, T.; Feldmann, H. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis. 2011, 204, 1075–1081. [Google Scholar]
- Falzarano, D.; Geisbert, T.W.; Feldmann, H. Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert. Rev. Vaccines. 2011, 10, 63–77. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jones, S.; Fritz, E.A. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005, 2, 537–545. [Google Scholar]
- Charrel, R.N.; de Lamballerie, X.; Fulhorst, C.F. The Whitewater Arroyo virus: natural evidence for genetic recombination among Tacaribe serocomplex viruses (family Arenaviridae). Virology 2001, 283, 161–166. [Google Scholar] [CrossRef]
- Riviere, Y.; Oldstone, M.B. Genetic reassortants of lymphocytic choriomeningitis virus: unexpected disease and mechanism of pathogenesis. J. Virol. 1986, 59, 363–368. [Google Scholar]
- Bishop, D.H.L.; Beaty, B.J.; Shope, R.E. Recombination and gene coding asignment of bunyaviruses and arenaviruses. Ann. N. Y. Acad. Sci. 1980, 354, 84–106. [Google Scholar] [CrossRef]
- Riviere, Y.R.; Ahmed, P.J.; Southern, M.J.; Buchmeier & Oldstone, M.B. Genetic mapping of lymphocytic choriomeningitis virus pathogenicity: virulence in guinea pigs is associated with the L RNA segment. J. Virol. 1985, 55, 704–709. [Google Scholar]
- Zhang, L.; Marriott, K.A.; Harnis, D.G.; Aronson, J.F. Reassortant analysis of guinea pig virulence of Pichinde virus variants. Virology 2001, 290, 30–38. [Google Scholar]
- Moshkoff, D.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2007, 34, 169–176. [Google Scholar] [CrossRef]
- Albarino, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Erickson, B.R.; Nichol, S.T. Efficient rescue of recombinant Lassa virus reveals the influence of S segment non-coding regions on virus replication and virulence. J. Virol. 2011, 85, 4020–4024. [Google Scholar] [CrossRef]
- Hass, M.; Westerkofsky, M.; Muller, S.; Becker-Ziaja, B.; Busch, C.; Gunther, S. Mutational Analysis of the Lassa Virus Promoter. J. Virol. 2006, 80, 12414–12419. [Google Scholar] [CrossRef]
- Perez, M.; de la Torre, J.C. Characterization of the Genomic Promoter of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2003, 77, 1184–1194. [Google Scholar]
- Barr, J.N.; Wertz, G.W. Bunyamwera Bunyavirus RNA Synthesis Requires Cooperation of 3'- and 5'-Terminal Sequences. J. Virol. 2004, 78, 1129–1138. [Google Scholar] [CrossRef]
- Lukashevich, I.; Djavani, M.; Shapiro, K. The Lassa fever virus L gene: nucleotide sequence, comparison, and precipitation of a predicted 250 kDa protein with monospecific antiserum. J. Gen. Virol. 1997, 78, 547–551. [Google Scholar]
- Lelke, M.; Brunotte, L.; Busch, C.; Gunther, S. An N-Terminal Region of Lassa Virus L Protein Plays a Critical Role in Transcription but Not Replication of the Virus Genome. J. Virol. 2010, 84, 1934–1944. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 2396–2401. [Google Scholar]
- Qi, X.; Lan, S.; Wang, W. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Identification of Amino Acid Residues Critical for the Anti-Interferon Activity of the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de La Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the Type I Interferon Response by the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Hastie, K.M.; Bale, S.; Kimberlin, C.R.; Saphire, E.O. Hiding the evidence: two strategies for innate immune evasion by hemorrhagic fever viruses. Curr. Opin. Virol. 2012, 2, 151–156. [Google Scholar]
- Glushakova, S.E.; Omelyanenko, V.G.; Lukashevitch, I.S. The fusion of artificial lipid membranes induced by the synthetic arenavirus 'fusion peptide'. Biochim. Biophys. Acta. 1992, 1110, 202–208. [Google Scholar] [CrossRef]
- Klewitz, C.; Klenk, H.-D.; ter Meulen, J. Amino acids from both N-terminal hydrophobic regions of the Lassa virus envelope glycoprotein GP-2 are critical for pH-dependent membrane fusion and infectivity. J. Gen. Virol. 2007, 88, 2320–2328. [Google Scholar]
- Moshkoff, D.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2006. [Google Scholar]
- Kelley, L.A.; Michael, J.E.S. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols 2009, 4, 363–371. [Google Scholar]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E. Substitutions in the Glycoprotein (GP) of the Candid#1 Vaccine Strain of Junin Virus Increase Dependence on Human Transferrin Receptor 1 for Entry and Destabilize the Metastable Conformation of GP. Journal of Virology 2011, 85, 13457–13462. [Google Scholar]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K. The Major Determinant of Attenuation in Mice of the Candid1 Vaccine for Argentine Hemorrhagic Fever Is Located in the G2 Glycoprotein Transmembrane Domain. Journal of Virology 2011, 85, 10404–10408. [Google Scholar] [CrossRef]
- Glushakova, S.E.; Pyzhik, E.V.; Vasiuchkov, A.D.; Erokhina, I.R.; Mar'iankova, R.F. pH-dependent fusion of eukaryotic cells, caused by arenaviruses, pathogenic and non-pathogenic for humans. Mol Gen Mikrobiol Virusol. 1992, 7-8, 27–31. [Google Scholar]
- Snoy, P.J. Establishing Efficacy of Human Products Using Animals. Veterinary Pathology Online 2010, 47, 774–778. [Google Scholar] [CrossRef]
- Goicocheaa, M.A.; Zapata, J.C.; Bryant, J.; Davis, H.; Salvato, M.S.; Lukashevich, I.S. Evaluation of Lassa virus vaccine immunogenicity in a CBA/J-ML29 mouse model. Vaccine 2012, 30, 1445–1452. [Google Scholar] [CrossRef]
- Barkar, N.D.; Lukashevich, I.S. Lassa and Mozambique viruses: cross protection in experiments on mice and action of immunosuppressants on experimental infections. Vopr. Virusol. 1989, 34, 598–603. [Google Scholar]
- Carrion, R.J.; Patterson, J.L.; Johnson, C. A ML29 reassortant virus protects guinea pigs against a distantly-related Nigerian strain of Lassa virus and can provide sterilizing immunity. Vaccine 2007, 25, 4093–4102. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008. In Press, Corrected Proof.. [Google Scholar]
- Zapata, J.C.; Poonia, B.; Bryant, J.; Davis, H.; Ateh, E.; George, L.; Crasta, O.; Zhang, Y.; Slezak, T.; Crystal Jaing, C.; Pauza, D.; Goicochea, M.; Moshkoff, D.; Lukashevich, I.S.; Salvato, M.S. An attenuated Lassa vaccine in SIV-infected rhesus macaques does not persist or cause arenavirus disease but does elicit Lassa virus-specific immunity. Virology J 2012. submitted. [Google Scholar]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar] [CrossRef]
- Hayes, E.B. Acute viscerotropic disease following vaccination against yellow fever. Trans R Soc Trop. Med. Hyg. 2007, 101, 967–971. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Schroeder, B.A.; Miller, E.R. Adverse event reports following yellow fever vaccination. Vaccine 2008, 26, 6077–6082. [Google Scholar]
- Silva, M.L.; Espirito-Santo, L.R.; Martins, M.A. Clinical and Immunological Insights on Severe, Adverse Neurotropic and Viscerotropic Disease following 17D Yellow Fever Vaccination. Clin. Vaccine Immunol. 2010, 17, 118–126. [Google Scholar]
- Galbraith, S.E.; Barrett, A.D.T. Vaccines for Biodefense and Emerging and Neglected Diseases. In Yellow Fever; Barrett, A.D.T., Stanberry, L.R., Eds.; Academic Press: Elsevier, Amsterdam, The Netherlands, 2009; pp. 753–785. [Google Scholar]
- Fisher-Hoch, S.; McCormick, J. Towards a human Lassa fever vaccine. Rev. Med. Virol. 2001, 11, 331–341. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- Barba-Spaeth, G.; Longman, R.S.; Albert, M.L.; Rice, C.M. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J. Exp. Med. 2005, 202, 1179–1184. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Garratt, R.C.; Caufour, P.S. Surface expression of an immunodominant malaria protein B cell epitope by yellow fever virus. J. Mol. Biol. 2002, 315, 873–885. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Garratt, R.C.; Marchevsky, R.S. Attenuation of Recombinant Yellow Fever 17D Viruses Expressing Foreign Protein Epitopes at the Surface. J. Virol. 2005, 79, 8602–8613. [Google Scholar]
- Tao, D.; Barba-Spaeth, G.; Rai, U.; Nussenzweig, V.; Rice, C.M.; Nussenzweig, R.S. Yellow fever 17D as a vaccine vector for microbial CTL epitopes: protection in a rodent malaria model. J. Exp. Med. 2005, 201, 201–209. [Google Scholar]
- Stoyanov, C.T.; Boscardin, S.B.; Deroubaix, S. Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 2010. In Press, Corrected Proof. [Google Scholar]
- Editorial. YF17D vector for Lassa fever vaccine. International Innovation 2010, 77–79.
- Bonaldo, M.; Mello, S.; Trindade, G. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virology Journal 2007, 4, 115. [Google Scholar]
- Bonaldo, M.C.; Martins, M.A.; Rudersdorf, R. Recombinant Yellow Fever Vaccine Virus 17D Expressing Simian Immunodeficiency Virus SIVmac239 Gag Induces SIV-Specific CD8+ T-Cell Responses in Rhesus Macaques. J. Virol. 2010, 84, 3699–3706. [Google Scholar]
- Stoyanov, C.T.; Boscardin, S.B.; Deroubaix, S. Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 2010, 28, 4644–4652. [Google Scholar] [CrossRef]
- Barban, V.; Girerd, Y.; Aguirre, M. High stability of yellow fever 17D-204 vaccine: A 12-year restrospective analysis of large-scale production. Vaccine 2007, 25, 2941–2950. [Google Scholar]
- Monath, T.P.; Myers, G.A.; Beck, R.A. Safety testing for neurovirulence of novel live, attenuated flavivirus vaccines: Infant mice provide an accurate surrogate for the test in monkeys. Biologicals 2005, 33, 131–144. [Google Scholar] [CrossRef]
- Monath, T.P.; Levenbook, I.; Soike, K. Chimeric Yellow Fever Virus 17D-Japanese Encephalitis Virus Vaccine: Dose-Response Effectiveness and Extended Safety Testing in Rhesus Monkeys. J. Virol. 2000, 74, 1742–1751. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 1987, 155, 465–467. [Google Scholar] [CrossRef]
- Peters, C.J. Arenaviruses; Richman, D.D., Whitley, R.J., Eds.; Churchill Livingstone: New York, 1997; pp. 779–799. [Google Scholar]
- Walker, D.H.; Wulff, H.; Lange, J.V.; Murphy, F.A. Comparative pathology of Lassa virus infection in monkeys, guinea-pigs, and Mastomys natalensis. Bull. World Health Organ. 1975, 52, 523–534. [Google Scholar]
- Hensley, L.E.; Smith, M.A.; Geisbert, J.B. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar]
- Carrion, R.J.; Patterson, J.L. Vaccines against viral hemorrhagic fevers: non-human primate models. Hum. Vaccin. 2011, 7, 667–673. [Google Scholar] [CrossRef]
- Mansfield, K. Marmoset model commonly used in biomedical research. Comp. Medicine 2003, 53, 383–392. [Google Scholar]
- Gowen, B.B.; Holbrook, M.R. Animal models of highly pathogenic RNA viral infections: Hemorrhagic fever viruses. Antivir. Res. 2008, 78, 79–90. [Google Scholar]
- Omatsu, T.; Moi, M.L.; Hirayama, T.; Takasaki, T.; Nakamura, S.; Tajima, S.; Ito, M.; Yoshida, T.; Saito, A.; Katakai, Y.; Akari, H.; Kurane, I. Common marmoset (Callithrix jacchus) as a primate model of dengue virus infection: development of high levels of viraemia and demonstration of protective immunity. J. Gen. Virol. 2011, 92, 2272–2280. [Google Scholar] [CrossRef]
- Firsova, I.; Shatokhina, I.V.; Borisevich, I.V.; Evseev, A.A.; Maksimov, V.A.; Pantiukhov, V.B.; Khmelev, A.L. Use of guinea pigs for assessing the efficacy of vaccines against Lassa fever. Vopr Virusol. 2003, 48, 43–45. [Google Scholar]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W. T cell-dependence of Lassa fever pathogenesis. PLoS Pathogen. 2010, 6, e1000836. [Google Scholar]
- Yun, N.E.; Poussard, A.L.; Seregin, A.V.; Walker, A.G.; Smith, J.K.; Aronson, J.F.; Smith, J.N.; Soong, L.; Paessler, S. Functional Interferon System Is Required for Clearance of Lassa Virus. J. Virol. 2012, 86, 3389–3392. [Google Scholar]
- Schildknecht, A.; Welti, S.; Geuking, M.B.; Hangartner, L.; van den Broek, M. Absence of CTL Responses to Early Viral Antigens Facilitates Viral Persistence. J. Immunol. 2008, 180, 3113–3121. [Google Scholar]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce robust type I IFN response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef]
- Marzi, A.; Feldmann, H.; Geisbert, T.W.; Falzarano, D. Vesicular Stomatitis Virus-Based Vaccines for Prophylaxis and Treatment of Filovirus Infections. J. Bioterror. Biodef. 2011. [Google Scholar]
- Appaiahgari, M.B.S.V. Clinical development of IMOJEV ® - a recombinant Japanese encephalitis chimeric vaccine (JE-CV). Exp. Opin. Biol. Ther. 2012. [Google Scholar]
- Guy, B.; Barrere, B.; Malinowski, C.; Saville, M.; Teyssou, R.; Lang, J. From research to phase III: Preclinical, industrial and clinical development of the Sanofi Pasteur tetravalent dengue vaccine. Vaccine 2011, 29, 7229–7241. [Google Scholar]
- Leroy, E.M.; Gonzale, J.P.; Baize, S. Ebola and Marburg haemorrhagic fever viruses: major scientific advances, but a relatively minor public health threat for Africa. Clin. Microbiol. Infect. 2011, 17, 964–976. [Google Scholar]
- McKee, K.T.J.; Enria, D.A.; Barerra-Oro, J.G. Vaccines for Biodefense and Energing and Neglected Diseases. In Junin (Argentine Hemorrhagic Fever); Barett, A.D.T., Stanberry, L.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 537–550. [Google Scholar]
- Anonymous. Arboviruses and Related Zoonotic Viruses. 2007. Available online: www.cdc.gov/biosafety/publications/bmbl5/BMBL5_sect_VIII_f.pdf.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lukashevich, I.S. Advanced Vaccine Candidates for Lassa Fever. Viruses 2012, 4, 2514-2557. https://doi.org/10.3390/v4112514
Lukashevich IS. Advanced Vaccine Candidates for Lassa Fever. Viruses. 2012; 4(11):2514-2557. https://doi.org/10.3390/v4112514
Chicago/Turabian StyleLukashevich, Igor S. 2012. "Advanced Vaccine Candidates for Lassa Fever" Viruses 4, no. 11: 2514-2557. https://doi.org/10.3390/v4112514
APA StyleLukashevich, I. S. (2012). Advanced Vaccine Candidates for Lassa Fever. Viruses, 4(11), 2514-2557. https://doi.org/10.3390/v4112514