Insights into the Functions of a Prophage Recombination Directionality Factor


Abstract
:1. Introduction
2. Results and Discussion
2.1. Tester Strain and Random Mutagenesis of the torI Gene


| Plasmid a | Mutation | Substitution | RDF activity b | Anti-TorR activity c |
|---|---|---|---|---|
| pJFi | N/A | N/A | + + + + | + |
| pJFi-L5P |  | Leu5 → Pro | + + + + | − |
| pJFi-S9L |  | Ser9 → Leu | − | − |
| pJFi-D12Y-D35G |  | Asp12 → Tyr | − | − |
| pJFi-F15L |  | Phe15 → Leu | − | − |
| pJFi-I16V |  | Ile16 → Val | + + | − |
| pJFi-M17V |  | Met17 → Val | + + | − |
| pJFi-F22I |  | Phe22 → Ile | − | − |
| d pJFi-Y28F |  | Tyr28 → Phe | − | − |
| d pJFi-Y28S |  | Tyr28 → Ser | − | − |
| pJFi-P37L |  | Pro37 → Leu | + | − |
| pJFi-H43Y-C54R |  | His43 → Tyr | + | − |
| pJFi-R45STOP |  | Arg45 → STOP | − | − |
| d pJFi-R45Q |  | Arg45 → Gln | + | − |
| d pJFi-R45K |  | Arg45 → Lys | + + | − |
| pJFi-A46V |  | Ala46 → Val | − | − |
| pJFi-A46T |  | Ala46 → Thr | − | − |
| pJFi-W48R |  | Trp48 → Arg | − | − |
| pJFi-E55G |  | Gln55 → Gly | + + + + | − |
| pJFi-F56L |  | Phe56 → Leu | + + + + | − |
| d pETsI-L61S |  | Leu61 → Ser | ND | ND |
| d pETsI-R63C-A64S |  | Arg63 → Cys | ND | ND |
| pJFi-N65Y |  | Asn65 → Tyr | + + + + | − |
| pJFi+18 |  | +18 residues e | + | − |
| pJFi+24 |  | +24 residues f | + + + + | − |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
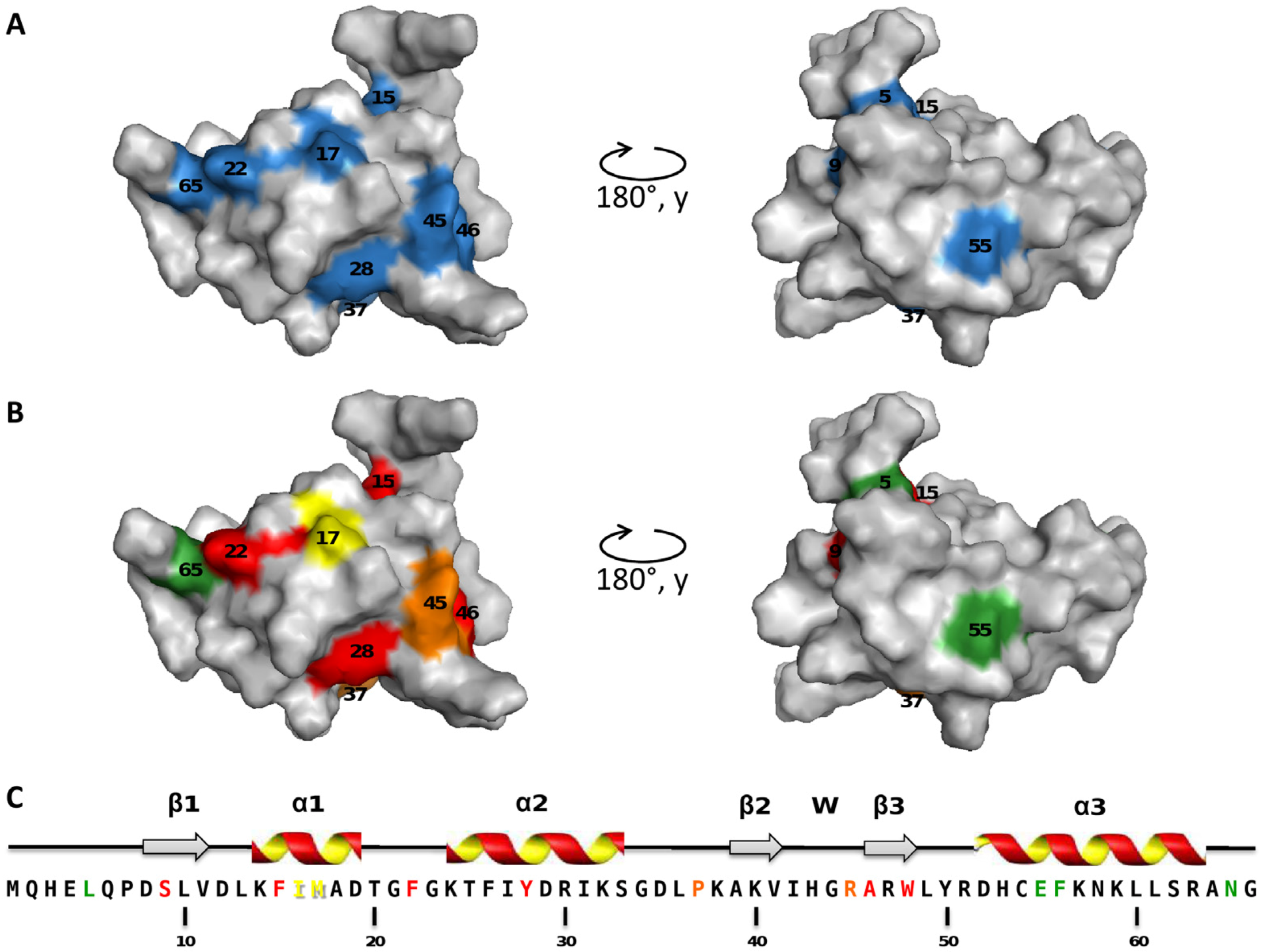
2.2. Mapping of the Mutations that Affect TorI Activities

2.3. Characterization of the TorI Mutants
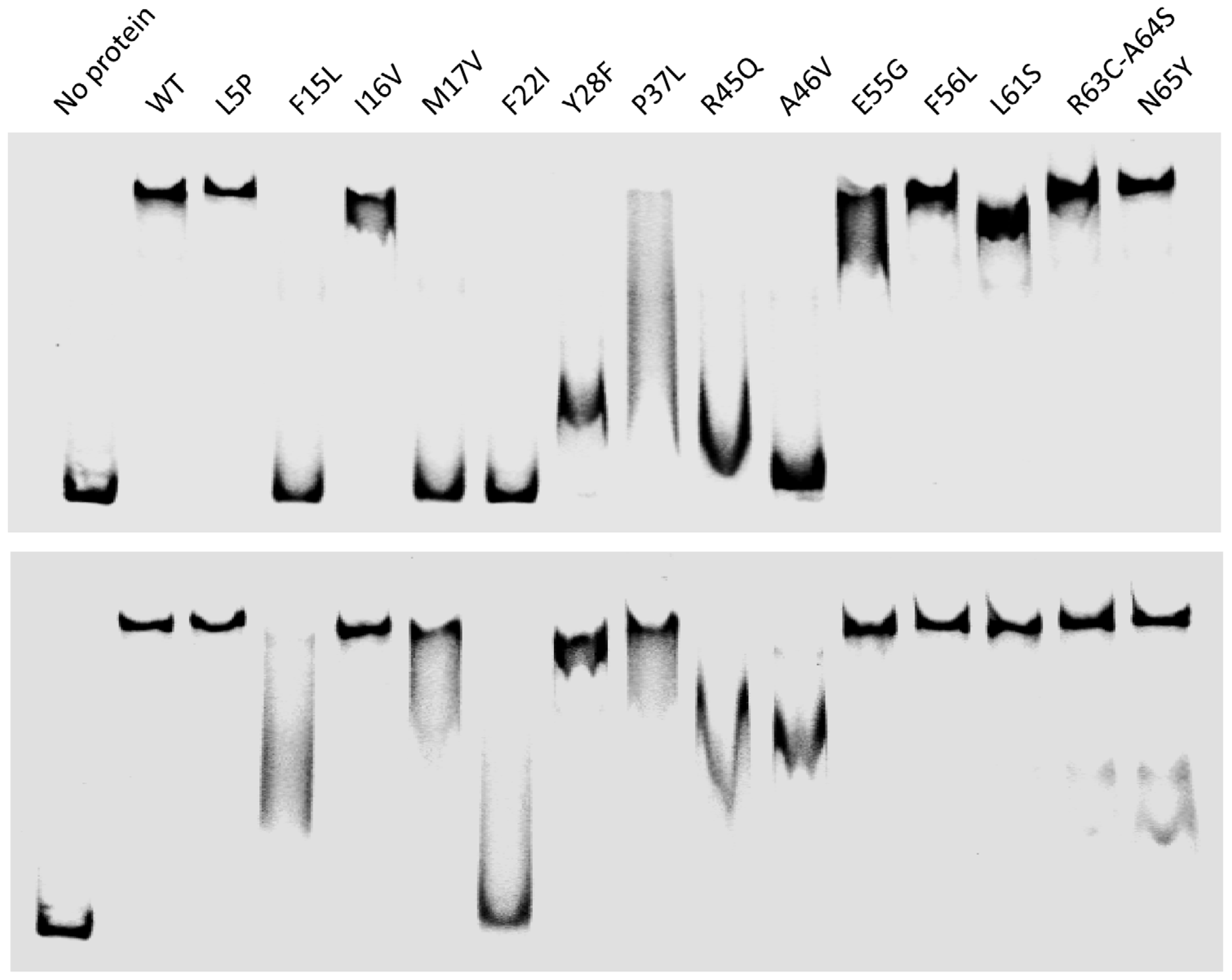
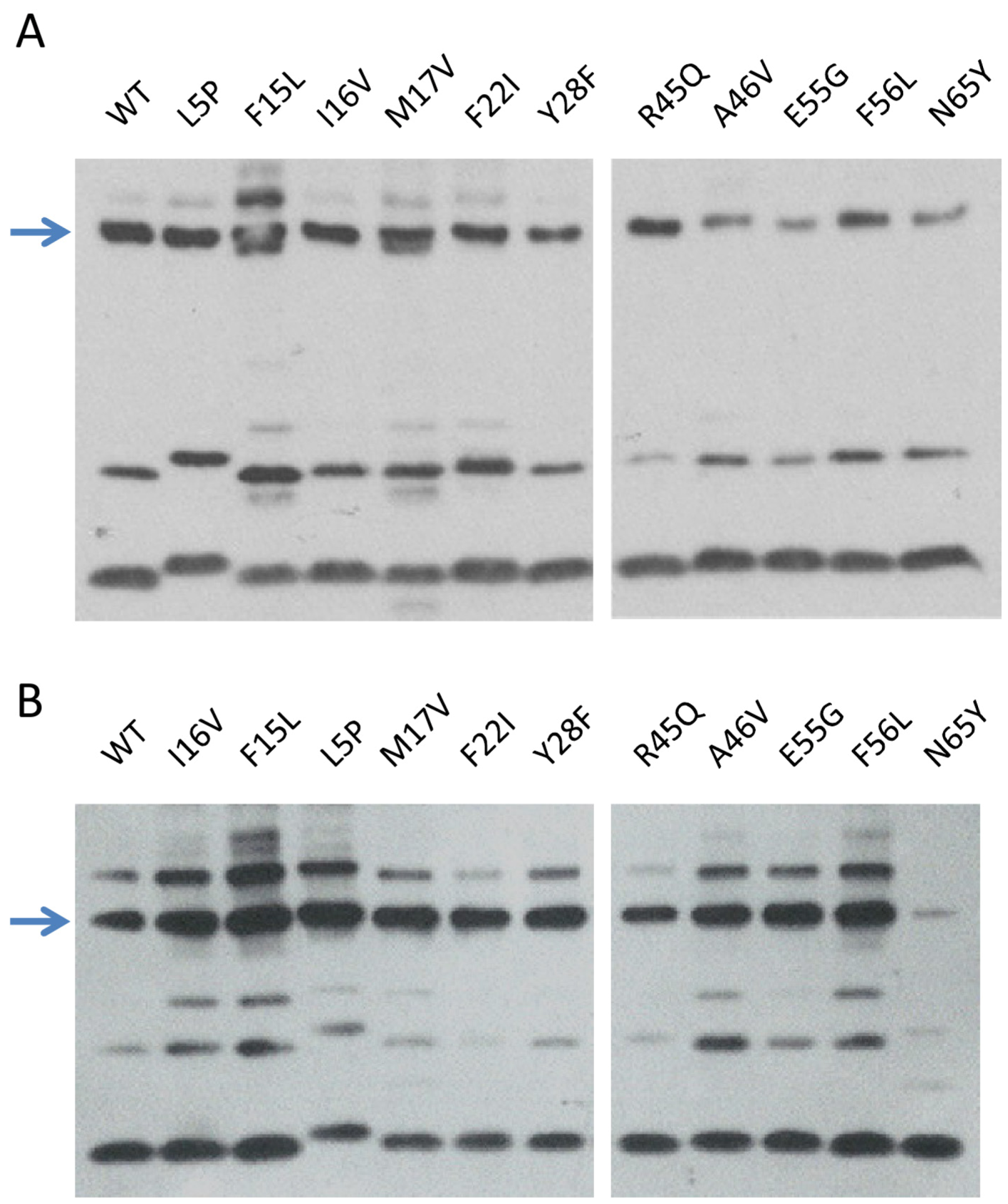
2.3.1. Production and Stability of the Mutants for in Vitro Studies
2.3.2. In Vitro RDF Activity of TorI Mutants

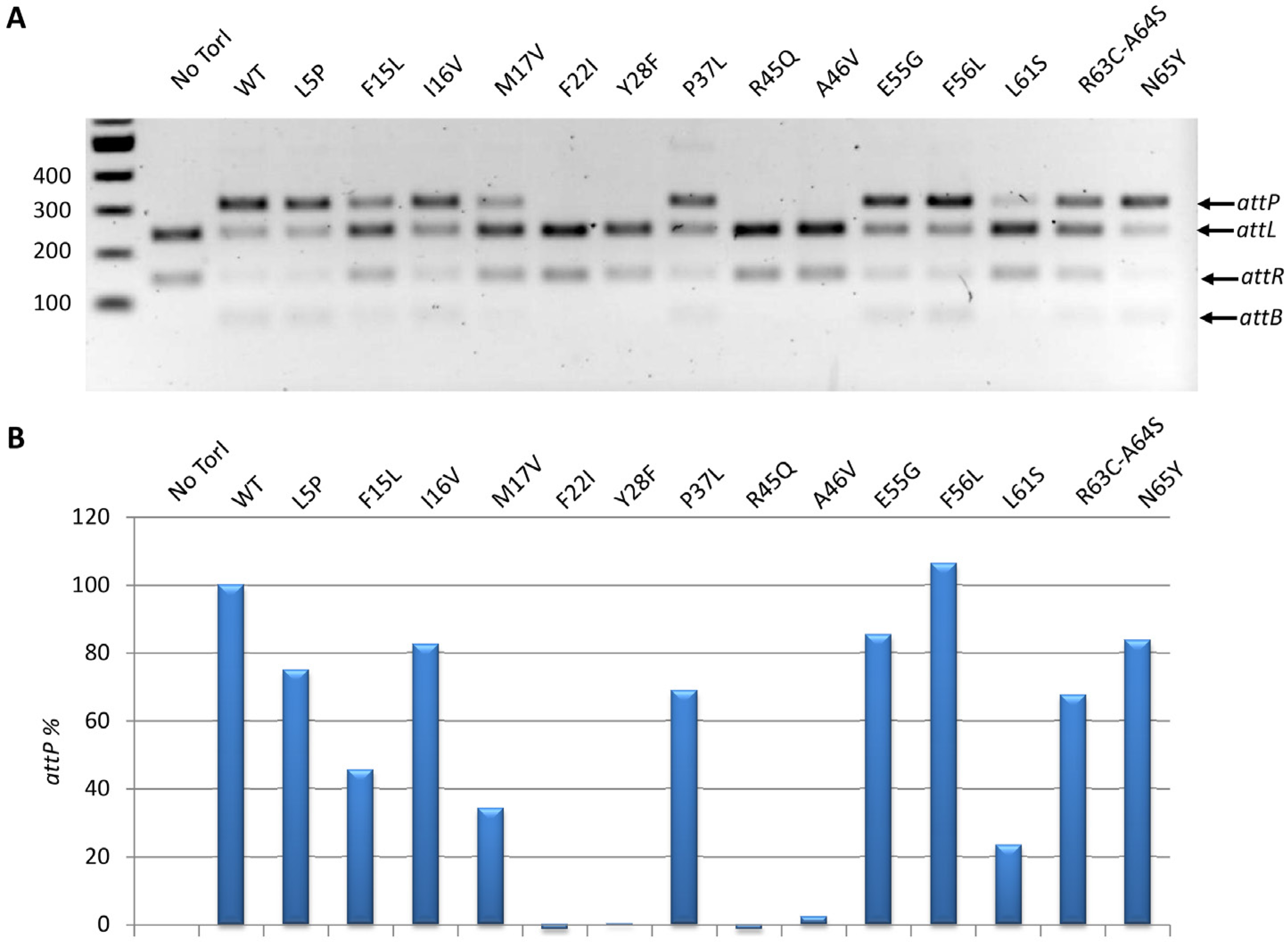
2.3.3. In Vitro Excisive Recombination

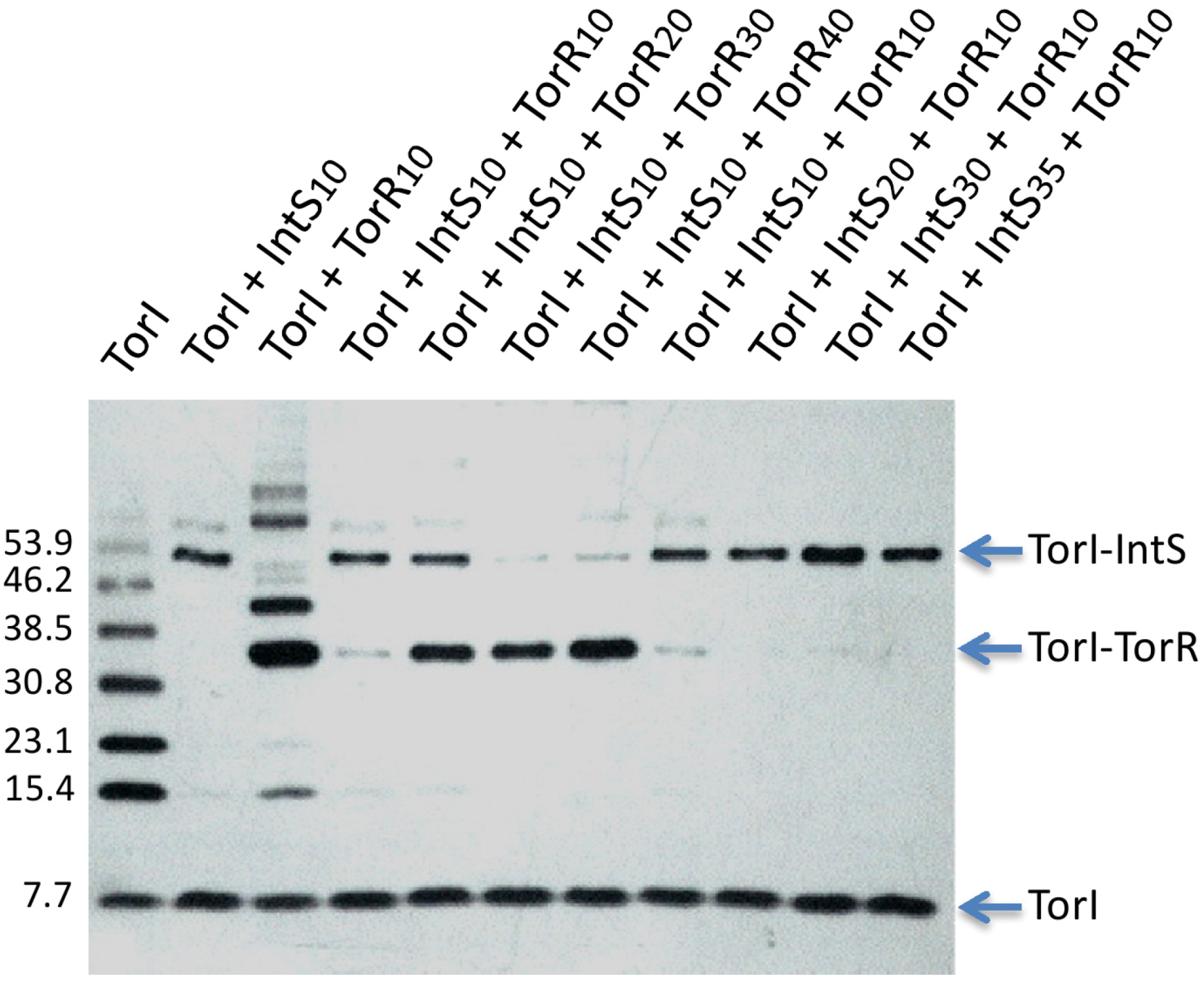
2.3.4. Competition for TorR and IntS Binding on TorI Protein

2.3.5. TorI Mutants Binding to TorR and IntS

3. Experimental Section
3.1. Strains and Media Used in This Study
| Strains and plasmids | Characteristics | Sources |
|---|---|---|
| Strains | ||
| MC4100 | araD139 (∆lacIPOZYA-argF) U169 rpsL thi | Casadaban |
| LCB620 | MC4100 torA8::MudII 1734 (torA'-'lacZ, KmR) | [27] |
| LCB970 | MC4100 yfdO-cat-yfdP | [24] |
| LCB995 | MC4100 torA8::MudII 1734, yfdO-cat-yfdP | This work |
| LCB984 | MC4100 yfdO-kan-yfdP | This work |
| BL21(DE3) | E. coli B F- [lon] dcm ompT hsdS (rB−mB+) gal λ(DE3) | Novagen |
| C41(DE3) | Derived from BL21(DE3) | [31] |
| Plasmids | ||
| pBAD33 | pACYC184 (ori p15A) vector containing a PBAD promoter (CmR) | [32] |
| pBtorR | torR coding sequence cloned into pBAD33 | [33] |
| pJF119EH | pBR322 (ori colE1) containing the IPTG inducible promoter ptac (ApR) | [34] |
| pJFi | torI coding sequence cloned into pJF119EH BamHI and EcoRI sites | [24] |
| pJFi* series | pJFi derived plasmids carrying torI mutated alleles | This work [20,26] |
| pET-22(+) | Promoter T7 containing vector (ApR) | Novagen |
| pETsi | torI coding sequence with a Stop codon cloned into pET-22(+) NdeI and XhoI sites | [24] |
| pETsi* series | pETsi derived plasmids carrying torI mutated alleles | This work [20,26] |
3.2. Random Mutagenesis
3.3. Protein Production and Purification
3.4. In Vivo Excision Assay
3.5. β-Galactosidase Assay
3.6. In Vitro Excisive Recombination
3.7. Electrophoretic Mobility Shift Assays (EMSA)
3.8. Cross-Linking Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Kourilsky, P.; Knapp, A. Lysogenization by bacteriophage lambda. III. Multiplicity dependent phenomena occuring upon infection by lambda. Biochimie 1974, 56, 1517–1523. [Google Scholar] [CrossRef]
- Edgar, R.; Rokney, A.; Feeney, M.; Semsey, S.; Kessel, M.; Goldberg, M.B.; Adhya, S.; Oppenheim, A.B. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. 2008, 68, 1107–1116. [Google Scholar] [CrossRef]
- Williamson, S.J.; Houchin, L.A.; McDaniel, L.; Paul, J.H. Seasonal variation in lysogeny as depicted by prophage induction in Tampa Bay, Florida. Appl. Environ. Microbiol. 2002, 68, 4307–4314. [Google Scholar] [CrossRef]
- McDaniel, L.; Paul, J.H. Effect of nutrient addition and environmental factors on prophage induction in natural populations of marine synechococcus species. Appl. Environ. Microbiol. 2005, 71, 842–850. [Google Scholar] [CrossRef]
- Rokney, A.; Kobiler, O.; Amir, A.; Court, D.L.; Stavans, J.; Adhya, S.; Oppenheim, A.B. Host responses influence on the induction of lambda prophage. Mol. Microbiol. 2008, 68, 29–36. [Google Scholar] [CrossRef]
- Nash, H. Site-specific recombination: Integration, excision, resolution, and inversion of defined DNA segments. In Escherichia coli and Salmonella: Cellular and Molecular Biology; Neidhardt, F., Ed.; ASM Press: Washington, DC, USA, 1996; pp. 2363–2376. [Google Scholar]
- Argos, P.; Landy, A.; Abremski, K.; Egan, J.B.; Haggard-Ljungquist, E.; Hoess, R.H.; Kahn, M.L.; Kalionis, B.; Narayana, S.V.; Pierson, L.S.; et al. The integrase family of site-specific recombinases: Regional similarities and global diversity. EMBO J. 1986, 5, 433–440. [Google Scholar]
- Nunes-Duby, S.E.; Kwon, H.J.; Tirumalai, R.S.; Ellenberger, T.; Landy, A. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 1998, 26, 391–406. [Google Scholar] [CrossRef]
- Gottesman, S.; Abremski, K. The role of HimA and Xis in lambda site-specific recombination. J. Mol. Biol. 1980, 138, 503–512. [Google Scholar] [CrossRef]
- Bushman, W.; Yin, S.; Thio, L.L.; Landy, A. Determinants of directionality in lambda site-specific recombination. Cell 1984, 39, 699–706. [Google Scholar] [CrossRef]
- Gardner, J.F.; Nash, H.A. Role of Escherichia coli IHF protein in lambda site-specific recombination. A mutational analysis of binding sites. J. Mol. Biol. 1986, 191, 181–189. [Google Scholar] [CrossRef]
- Lewis, J.A.; Hatfull, G.F. Control of directionality in integrase-mediated recombination: Examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res. 2001, 29, 2205–2216. [Google Scholar] [CrossRef]
- Numrych, T.E.; Gumport, R.I.; Gardner, J.F. Characterization of the bacteriophage lambda excisionase (Xis) protein: The C-terminus is required for Xis-integrase cooperativity but not for DNA binding. EMBO J. 1992, 11, 3797–3806. [Google Scholar]
- Sam, M.D.; Papagiannis, C.V.; Connolly, K.M.; Corselli, L.; Iwahara, J.; Lee, J.; Phillips, M.; Wojciak, J.M.; Johnson, R.C.; Clubb, R.T. Regulation of directionality in bacteriophage lambda site-specific recombination: Structure of the Xis protein. J. Mol. Biol. 2002, 324, 791–805. [Google Scholar] [CrossRef]
- Warren, D.; Sam, M.D.; Manley, K.; Sarkar, D.; Lee, S.Y.; Abbani, M.; Wojciak, J.M.; Clubb, R.T.; Landy, A. Identification of the lambda integrase surface that interacts with Xis reveals a residue that is also critical for Int dimer formation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8176–8181. [Google Scholar]
- Touchon, M.; Hoede, C.; Tenaillon, O.; Barbe, V.; Baeriswyl, S.; Bidet, P.; Bingen, E.; Bonacorsi, S.; Bouchier, C.; Bouvet, O.; et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009, 5, e1000344. [Google Scholar] [CrossRef] [Green Version]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brussow, H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Campbell, A.M. Cryptic prophages. In Escherichia coli and Salmonella: Cellular and Molecular Biology; Neidhardt, F., Ed.; ASM Press: Washington, DC, USA, 1996; pp. 2041–2046. [Google Scholar]
- ElAntak, L.; Ansaldi, M.; Guerlesquin, F.; Méjean, V.; Morelli, X. Structural and genetic analyses reveal a key role in prophage excision for the TorI response regulator inhibitor. J. Biol. Chem. 2005, 280, 36802–36808. [Google Scholar]
- Panis, G.; Méjean, V.; Ansaldi, M. Control and regulation of KplE1 prophage site-specific recombination: A new recombination module analyzed. J. Biol. Chem. 2007, 282, 21798–21809. [Google Scholar] [CrossRef]
- Clark, A.J.; Inwood, W.; Cloutier, T.; Dhillon, T.S. Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J. Mol. Biol. 2001, 311, 657–679. [Google Scholar] [CrossRef]
- Casjens, S.; Winn-Stapley, D.A.; Gilcrease, E.B.; Morona, R.; Kuhlewein, C.; Chua, J.E.; Manning, P.A.; Inwood, W.; Clark, A.J. The chromosome of Shigella flexneri bacteriophage Sf6: Complete nucleotide sequence, genetic mosaicism, and DNA packaging. J. Mol. Biol. 2004, 339, 379–394. [Google Scholar] [CrossRef]
- Ansaldi, M.; Théraulaz, L.; Méjean, V. TorI, a response regulator inhibitor of phage origin in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9423–9428. [Google Scholar] [CrossRef]
- Champ, S.; Puvirajesinghe, T.M.; Perrody, E.; Menouni, R.; Genevaux, P.; Ansaldi, M. Chaperone-assisted excisive recombination, a solitary role for DnaJ (Hsp40) chaperone in lysogeny escape. J. Biol. Chem. 2011, 286, 38876–38885. [Google Scholar]
- Puvirajesinghe, T.M.; Elantak, L.; Lignon, S.; Franche, N.; Ilbert, M.; Ansaldi, M. DnaJ (HSP40) binding to a folded substrate impacts KplE1 prophage excision efficiency. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Méjean, V.; Iobbi-Nivol, C.; Lepelletier, M.; Giordano, G.; Chippaux, M.; Pascal, M.C. TMAO anaerobic respiration in Escherichia coli: Involvement of the tor operon. Mol. Microbiol. 1994, 11, 1169–1179. [Google Scholar] [CrossRef]
- Martinez-Hackert, E.; Stock, A.M. The DNA-binding domain of OmpR: Crystal structures of a winged helix transcription factor. Structure 1997, 5, 109–124. [Google Scholar] [CrossRef]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.M.; Iyer, L.M. The many faces of thehelix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar]
- Panis, G.; Duverger, Y.; Champ, S.; Ansaldi, M. Protein binding sites involved in the assembly of the KplE1 prophage intasome. Virology 2010, 404, 41–50. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar]
- Bordi, C.; Ansaldi, M.; Gon, S.; Jourlin-Castelli, C.; Iobbi-Nivol, C.; Méjean, V. Genes regulated by TorR, the trimethylamine oxide response regulator of Shewanella oneidensis. J. Bacteriol. 2004, 186, 4502–4509. [Google Scholar] [CrossRef]
- Furste, J.P.; Pansegrau, W.; Frank, R.; Blocker, H.; Scholz, P.; Bagdasarian, M.; Lanka, E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 1986, 48, 119–131. [Google Scholar] [CrossRef]
- Multigauge, version 2.3, Fujifilm: Tokyo, Japan, 2004.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Panis, G.; Franche, N.; Méjean, V.; Ansaldi, M. Insights into the Functions of a Prophage Recombination Directionality Factor. Viruses 2012, 4, 2417-2431. https://doi.org/10.3390/v4112417
Panis G, Franche N, Méjean V, Ansaldi M. Insights into the Functions of a Prophage Recombination Directionality Factor. Viruses. 2012; 4(11):2417-2431. https://doi.org/10.3390/v4112417
Chicago/Turabian StylePanis, Gaël, Nathalie Franche, Vincent Méjean, and Mireille Ansaldi. 2012. "Insights into the Functions of a Prophage Recombination Directionality Factor" Viruses 4, no. 11: 2417-2431. https://doi.org/10.3390/v4112417
APA StylePanis, G., Franche, N., Méjean, V., & Ansaldi, M. (2012). Insights into the Functions of a Prophage Recombination Directionality Factor. Viruses, 4(11), 2417-2431. https://doi.org/10.3390/v4112417