Diversity and Adaptation of Human Respiratory Syncytial Virus Genotypes Circulating in Two Distinct Communities: Public Hospital and Day Care Center

Abstract
:1. Introduction
2. Results and Discussion
2.1 HRSV Epidemiology and Cohorts Characterization
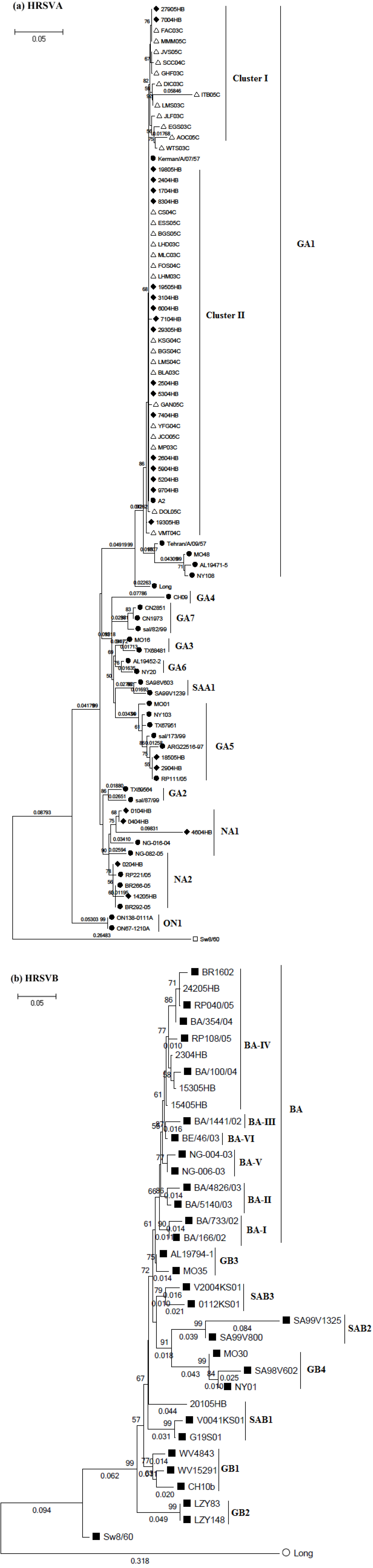
2.2 Phylogenetic Analysis
2.3 Molecular Analysis of HRSV GA1 Genotype
2.4 Selective Pressure

{kind=link}
{kind=link}
| HRSV | N | Π | D | d | p |
|---|---|---|---|---|---|
| A | 55 | 0.03465 | -2.14299 | 0.048 | p<0.05 |
| B | 5 | 0.03878 | -0.85185 | 0.040 | p>0.10 |
| GA1 | 48 | 0.00924 | -2.32187 | 0.010 | P<0.01 |


3. Experimental Procedures
3.1 Cohorts and Clinical specimens
3.2 Viral detection and Nucleotide Sequencing
3.3 Phylogenetic Analysis
3.4 O-glycosylation and N-glycosilation site analysis
3.5 Selection pressure analysis
3.6 Ethics Statement
Acknowledgments
Conflict of Interest
Supplementary Files
References
- Glezen, P.; Denny, F.W. Epidemiology of acute lower respiratory disease in children. N. Engl. J. Med. 1973, 288, 498–505. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; Grijalva, C.G.; Zhu, Y.; Szilagyi, P. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef]
- Anderson, L.J.; Parker, R.A.; Strikas, R.L. Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J. Infect. Dis. 1990, 161, 640–646. [Google Scholar] [CrossRef]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef]
- Widjojoatmodjo, M.N.; Boes, J.; van Bers, M.; van Remmerden, Y.; Roholl, P.J.; Luytjes, W. A highly attenuated recombinant human respiratory syncytial virus lacking the G protein induces long-lasting protection in cotton rats. Virol. J. 2010, 7, 114. [Google Scholar] [CrossRef]
- Cane, P.A. Molecular epidemiology of respiratory syncytial virus. Rev. Med. Virol. 2001, 11, 103–116. [Google Scholar] [CrossRef]
- Mufson, M.A.; Orvell, C.; Rafnar, B.; Norrby, E. Two distinct subtypes of human respiratory syncytial virus. J. Gen. Virol. 1985, 66 Pt 10, 2111–2124. [Google Scholar]
- Anderson, L.J.; Hierholzer, J.C.; Tsou, C.; Hendry, R.M.; Fernie, B.F.; Stone, Y.; McIntosh, K. Antigenic characterization of respiratory syncytial virus strains with monoclonal antibodies. J. Infect. Dis. 1985, 151, 626–633. [Google Scholar] [CrossRef]
- Peret, T.C.; Hall, C.B.; Schnabel, K.C.; Golub, J.A.; Anderson, L.J. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J. Gen. Virol. 1998, 79 Pt 9, 2221–2229. [Google Scholar]
- Peret, T.C.; Hall, C.B.; Hammond, G.W.; Piedra, P.A.; Storch, G.A.; Sullender, W.M.; Tsou, C.; Anderson, L.J. Circulation patterns of group A and B human respiratory syncytial virus genotypes in 5 communities in North America. J. Infect. Dis. 2000, 181, 1891–1896. [Google Scholar] [CrossRef]
- Venter, M.; Madhi, S.A.; Tiemessen, C.T.; Schoub, B.D. Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J. Gen. Virol. 2001, 82, 2117–2124. [Google Scholar]
- Shobugawa, Y.; Saito, R.; Sano, Y.; Zaraket, H.; Suzuki, Y.; Kumaki, A.; Dapat, I.; Oguma, T.; Yamaguchi, M.; Suzuki, H. Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J. Clin. Microbiol. 2009, 47, 2475–2482. [Google Scholar] [CrossRef]
- Eshaghi, A.; Duvvuri, V.R.; Lai, R.; Nadarajah, J.T.; Li, A.; Patel, S.N.; Low, D.E.; Gubbay, J.B. Genetic variability of human respiratory syncytial virus a strains circulating in ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 2012, 7, e32807. [Google Scholar]
- Trento, A.; Viegas, M.; Galiano, M.; Videla, C.; Carballal, G.; Mistchenko, A.S.; Melero, J.A. Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J. Virol. 2006, 80, 975–984. [Google Scholar] [CrossRef]
- Sullender, W.M. Respiratory syncytial virus genetic and antigenic diversity. Clin. Microbiol. Rev. 2000, 13, 1–15. [Google Scholar] [CrossRef]
- Zhang, R.F.; Jin, Y.; Xie, Z.P.; Liu, N.; Yan, K.L.; Gao, H.C.; Song, J.R.; Yuan, X.H.; Xiao, N.G.; Guo, M.W.; Zhou, Q.H.; Hou, Y.D.; Duan, Z. Human respiratory syncytial virus in children with acute respiratory tract infections in China. J. Clin. Microbiol. 2010, 48, 4193–4199. [Google Scholar] [CrossRef]
- Johnson, P.R.; Spriggs, M.K.; Olmsted, R.A.; Collins, P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 5625–5629. [Google Scholar]
- Garcia-Beato, R.; Martinez, I.; Franci, C.; Real, F.X.; Garcia-Barreno, B.; Melero, J.A. Host cell effect upon glycosylation and antigenicity of human respiratory syncytial virus G glycoprotein. Virology 1996, 221, 301–309. [Google Scholar] [CrossRef]
- Lambert, D.M. Role of oligosaccharides in the structure and function of respiratory syncytial virus glycoproteins. Virology 1988, 164, 458–466. [Google Scholar] [CrossRef]
- Cintra, O.A.; Owa, M.A.; Machado, A.A.; Cervi, M.C.; Figueiredo, L.T.; Rocha, G.M.; Siqueira, M.M.; Arruda, E. Occurrence and severity of infections caused by subgroup A and B respiratory syncytial virus in children in southeast Brazil. J. Med. Virol. 2001, 65, 408–412. [Google Scholar] [CrossRef]
- Moura, F.E.; Blanc, A.; Frabasile, S.; Delfraro, A.; de Sierra, M.J.; Tome, L.; Ramos, E.A.; Siqueira, M.M.; Arbiza, J. Genetic diversity of respiratory syncytial virus isolated during an epidemic period from children of northeastern Brazil. J. Med. Virol. 2004, 74, 156–160. [Google Scholar]
- Oliveira, T.F.; Freitas, G.R.; Ribeiro, L.Z.; Yokosawa, J.; Siqueira, M.M.; Portes, S.A.; Silveira, H.L.; Calegari, T.; Costa, L.F.; Mantese, O.C.; Queiroz, D.A. Prevalence and clinical aspects of respiratory syncytial virus A and B groups in children seen at Hospital de Clinicas of Uberlandia, MG, Brazil. Mem. Inst. Oswaldo Cruz 2008, 103, 417–422. [Google Scholar] [CrossRef]
- da Silva, L.H.; Spilki, F.R.; Riccetto, A.G.; de Almeida, R.S.; Baracat, E.C.; Arns, C.W. Genetic variability in the G protein gene of human respiratory syncytial virus isolated from the Campinas metropolitan region, Brazil. J. Med. Virol. 2008, 80, 1653–1660. [Google Scholar] [CrossRef]
- Riccetto, A.G.; Silva, L.H.; Spilki, F.R.; Morcillo, A.M.; Arns, C.W.; Baracat, E.C. Genotypes and clinical data of respiratory syncytial virus and metapneumovirus in brazilian infants: a new perspective. Braz. J. Infect. Dis. 2009, 13, 35–39. [Google Scholar] [CrossRef]
- Botosso, V.F.; Zanotto, P.M.; Ueda, M.; Arruda, E.; Gilio, A.E.; Vieira, S.E.; Stewien, K.E.; Peret, T.C.; Jamal, L.F.; Pardini, M.I.; Pinho, J.R.; Massad, E.; Sant'anna, O.A.; Holmes, E.C.; Durigon, E.L. Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog. 2009, 5, e1000254. [Google Scholar]
- Lima, H.N.; Botosso, V.F.; Oliveira, D.B.; Campos, A.C.; Leal, A.L.; Silva, T.S.; Bosso, P.A.; Moraes, C.T.; Filho, C.G.; Vieira, S.E.; Gilio, A.E.; Stewien, K.E.; Durigon, E.L. Molecular epidemiology of the SH (small hydrophobic) gene of human respiratory syncytial virus (HRSV), over 2 consecutive years. Virus. Res. 2012, 163, 82–86. [Google Scholar] [CrossRef]
- McCutcheon, H.; Fitzgerald, M. The public health problem of acute respiratory illness in childcare. J. Clin. Nurs. 2001, 10, 305–310. [Google Scholar] [CrossRef]
- Lu, N.; Samuels, M.E.; Shi, L.; Baker, S.L.; Glover, S.H.; Sanders, J.M. Child day care risks of common infectious diseases revisited. Child Care Health Dev. 2004, 30, 361–368. [Google Scholar] [CrossRef]
- Gardinassi, L.G.A.; Simas, P.V.M.; Salomão, J.B.; Durigon, E.L.; Trevisan, D.M.Z.; Cordeiro, J.A.; Lacerda, M.N.; Rahal, P.; Souza, F.P. Seasonality of viral respiratory infections in Southeast of Brazil: The influence of temperature and air humidity. Braz. J. of Microbiol. 2012, 43, 98–108. [Google Scholar] [CrossRef]
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef]
- Hall, C.B.; Walsh, E.E.; Schnabel, K.C.; Long, C.E.; McConnochie, K.M.; Hildreth, S.W.; Anderson, L.J. Occurrence of groups A and B of respiratory syncytial virus over 15 years: associated epidemiologic and clinical characteristics in hospitalized and ambulatory children. J. Infect. Dis. 1990, 162, 1283–1290. [Google Scholar] [CrossRef]
- Mufson, M.A.; Belshe, R.B.; Orvell, C.; Norrby, E. Respiratory syncytial virus epidemics: variable dominance of subgroups A and B strains among children, 1981–1986. J Infect Dis 1988, 157, 143–148. [Google Scholar] [CrossRef]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Rahman, M.; Van Ranst, M. Loop model: mechanism to explain partial gene duplications in segmented dsRNA viruses. Biochem. Biophys. Res. Commun. 2006, 340, 140–144. [Google Scholar] [CrossRef]
- Ballard, A.; McCrae, M.A.; Desselberger, U. Nucleotide sequences of normal and rearranged RNA segments 10 of human rotaviruses. J. Gen. Virol. 1992, 73 Pt 3, 633–638. [Google Scholar]
- Kojima, K.; Taniguchi, K.; Kawagishi-Kobayashi, M.; Matsuno, S.; Urasawa, S. Rearrangement generated in double genes, NSP1 and NSP3, of viable progenies from a human rotavirus strain. Virus Res. 2000, 67, 163–171. [Google Scholar] [CrossRef]
- Collins, P.L.; Chanock, R.M.; Murphy, B.R. Respiratory syncytial virus. In Fields Virology, 4th; Howley, D.M.K.P.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2001; pp. 1443–1485. [Google Scholar]
- Zlateva, K.T.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup a: positively selected sites in the attachment g glycoprotein. J. Virol. 2004, 78, 4675–4683. [Google Scholar] [CrossRef]
- Zlateva, K.T.; Lemey, P.; Moes, E.; Vandamme, A.M.; Van Ranst, M. Genetic variability and molecular evolution of the human respiratory syncytial virus subgroup B attachment G protein. J. Virol. 2005, 79, 9157–9167. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar]
- Gaunt, E.R.; Jansen, R.R.; Poovorawan, Y.; Templeton, K.E.; Toms, G.L.; Simmonds, P. Molecular epidemiology and evolution of human respiratory syncytial virus and human metapneumovirus. PLoS One 2011, 6, e17427. [Google Scholar]
- Garcia, O.; Martin, M.; Dopazo, J.; Arbiza, J.; Frabasile, S.; Russi, J.; Hortal, M.; Perez-Brena, P.; Martinez, I.; Garcia-Barreno, B.; et al. Evolutionary pattern of human respiratory syncytial virus (subgroup A): cocirculating lineages and correlation of genetic and antigenic changes in the G glycoprotein. J. Virol. 1994, 68, 5448–5459. [Google Scholar]
- Rueda, P.; Delgado, T.; Portela, A.; Melero, J.A.; Garcia-Barreno, B. Premature stop codons in the G glycoprotein of human respiratory syncytial viruses resistant to neutralization by monoclonal antibodies. J. Virol. 1991, 65, 3374–3378. [Google Scholar]
- Zheng, H.; Peret, T.C.; Randolph, V.B.; Crowley, J.C.; Anderson, L.J. Strain-specific reverse transcriptase PCR assay: means to distinguish candidate vaccine from wild-type strains of respiratory syncytial virus. J. Clin. Microbiol. 1996, 34, 334–337. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony method. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Julenius, K.; Molgaard, A.; Gupta, R.; Brunak, S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 2005, 15, 153–164. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gardinassi, L.G.A.; Simas, P.V.M.; Gomes, D.E.; Bonfim, C.M.d.; Nogueira, F.C.; Garcia, G.R.; Carareto, C.M.A.; Rahal, P.; Souza, F.P.d. Diversity and Adaptation of Human Respiratory Syncytial Virus Genotypes Circulating in Two Distinct Communities: Public Hospital and Day Care Center. Viruses 2012, 4, 2432-2447. https://doi.org/10.3390/v4112432
Gardinassi LGA, Simas PVM, Gomes DE, Bonfim CMd, Nogueira FC, Garcia GR, Carareto CMA, Rahal P, Souza FPd. Diversity and Adaptation of Human Respiratory Syncytial Virus Genotypes Circulating in Two Distinct Communities: Public Hospital and Day Care Center. Viruses. 2012; 4(11):2432-2447. https://doi.org/10.3390/v4112432
Chicago/Turabian StyleGardinassi, Luiz Gustavo Araujo, Paulo Vitor Marques Simas, Deriane Elias Gomes, Caroline Measso do Bonfim, Felipe Cavassan Nogueira, Gustavo Rocha Garcia, Claudia Márcia Aparecida Carareto, Paula Rahal, and Fátima Pereira de Souza. 2012. "Diversity and Adaptation of Human Respiratory Syncytial Virus Genotypes Circulating in Two Distinct Communities: Public Hospital and Day Care Center" Viruses 4, no. 11: 2432-2447. https://doi.org/10.3390/v4112432
APA StyleGardinassi, L. G. A., Simas, P. V. M., Gomes, D. E., Bonfim, C. M. d., Nogueira, F. C., Garcia, G. R., Carareto, C. M. A., Rahal, P., & Souza, F. P. d. (2012). Diversity and Adaptation of Human Respiratory Syncytial Virus Genotypes Circulating in Two Distinct Communities: Public Hospital and Day Care Center. Viruses, 4(11), 2432-2447. https://doi.org/10.3390/v4112432