The Staphylococci Phages Family: An Overview

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Phages of S. aureus
2.1. Global Features of S. aureus Phages
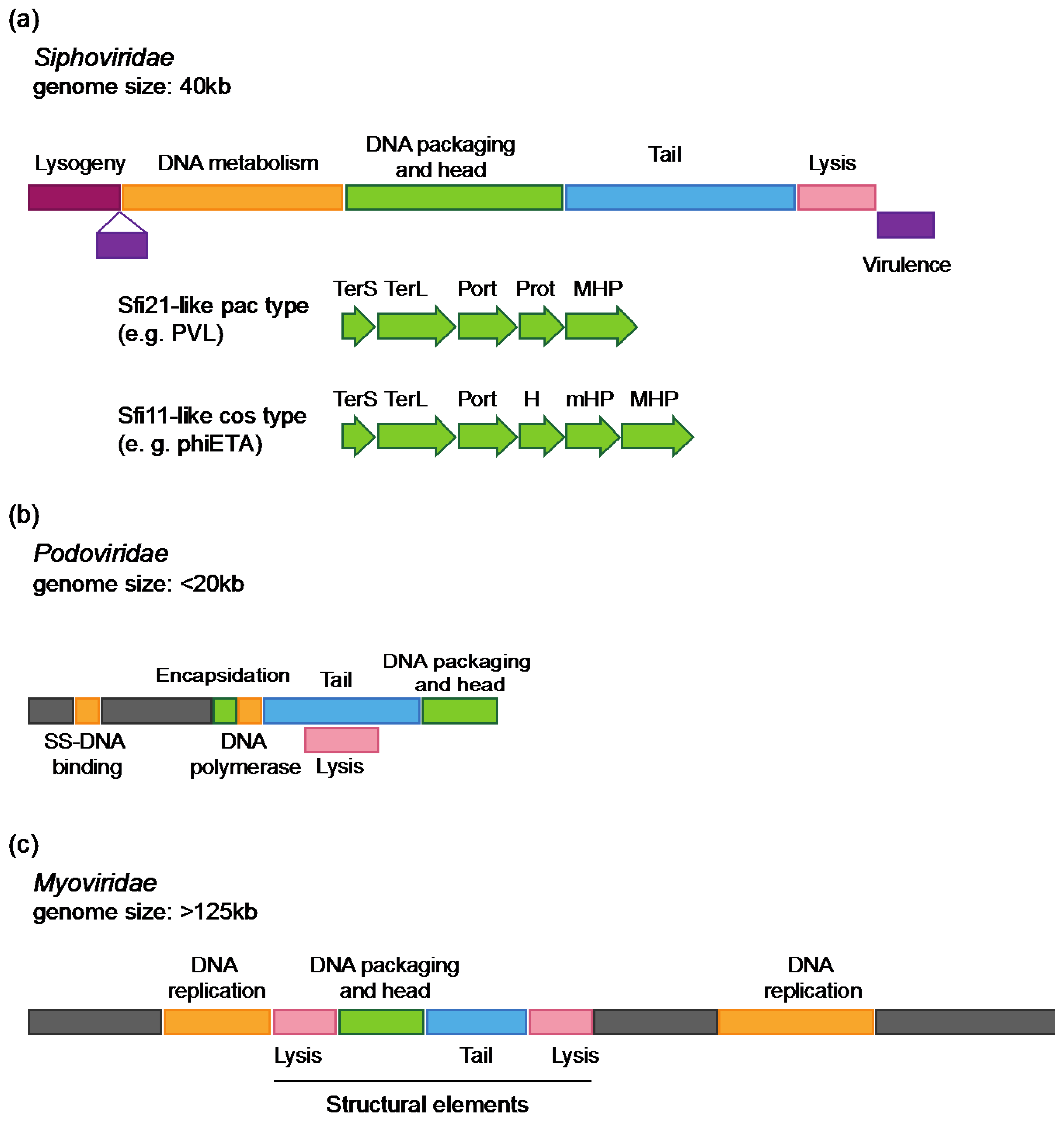
2.1.1. Morphological Families
2.1.2. Genomic Characteristics of S. aureus Phages

2.2. Role of Phages in S. aureus Pathogenesis
2.2.1. Phages-encoded Virulence Factors
2.2.2. Phage-mediated Mobilization of Virulence Factors: SaPIs Pathogenicity Islands
2.2.3. Phage Dynamics Contribute to S. aureus Evolution and Pathogenesis
3. What About Phages in Non-aureus Staphylococci?
4. Classification and Evolution of the Staphylococcal Phage Family
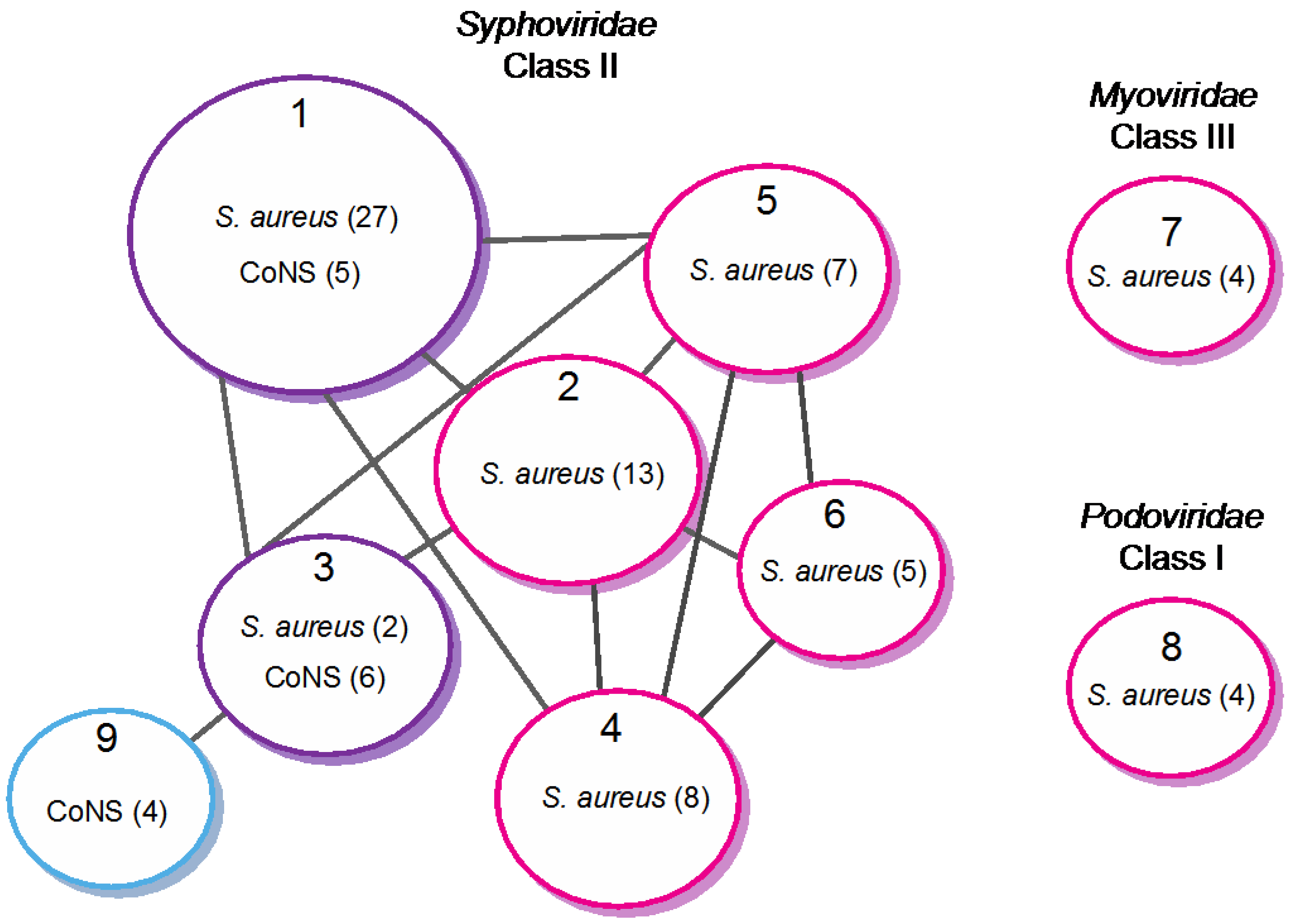
4.1. Classification of Staphylococci Phages, a Long-Term Challenge
4.2. Modular Evolution of Staphylococci Phages
4.2.1. Extensive Genome Mosaicism in Staphylococci Phages

4.2.2. Evolutionary Relationships within Staphylococci Phages
4.3. Evolutionary Relationships between Staphylococci Phages and Other Species
5. Use of Staphylococci Phages for Phage Therapy and Other Bio-Technological Applications
6. Concluding Remarks
Acknowledgments
Conflict of Interest
Supplementary Files
Supplementary File 1References
- Lindsay, J.A. Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 98–103. [Google Scholar] [CrossRef]
- Brussow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brussow, H. Prophage Genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef]
- Goerke, C.; Wirtz, C.; Fluckiger, U.; Wolz, C. Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 2006, 61, 1673–1685. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, C.J.; Su, L.H.; Hu, S.; Yu, J.; Chiu, C.H. Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics. FEMS Microbiol. Rev. 2008, 32, 23–37. [Google Scholar] [CrossRef]
- Wentworth, B.B. Bacteriophage Typing of the Staphylococci. Bacteriol. Rev. 1963, 27, 253–272. [Google Scholar]
- Rosenblum, E.D.; Tyrone, S. Serology, Density, and Morphology of Staphylococcal Phages. J. Bacteriol. 1964, 88, 1737–1742. [Google Scholar]
- Otto, M. Molecular basis of Staphylococcus epidermidis infections. Semin. Immunopathol. 2012, 34, 201–214. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus epidermidis--the 'accidental' pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef]
- von Eiff, C.; Peters, G.; Heilmann, C. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect. Dis. 2002, 2, 677–685. [Google Scholar] [CrossRef]
- Frank, K.L.; Del Pozo, J.L.; Patel, R. From clinical microbiology to infection pathogenesis: how daring to be different works for Staphylococcus lugdunensis. Clin. Microbiol. Rev. 2008, 21, 111–133. [Google Scholar] [CrossRef]
- Kuroda, M.; Yamashita, A.; Hirakawa, H.; Kumano, M.; Morikawa, K.; Higashide, M.; Maruyama, A.; Inose, Y.; Matoba, K.; Toh, H.; Kuhara, S.; Hattori, M.; Ohta, T. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc. Natl. Acad. Sci. USA 2005, 102, 13272–13277. [Google Scholar]
- EMBL phage database. Genomes pages - Phages. Available online: http://www.ebi.ac.uk/genomes/phage.html (accessed on 30 October 2012).
- Patric database. Staphylococcus page. Available online: http://www.patricbrc.org/portal/portal/patric/Taxon?cType=taxon&cId=1279 (accessed on accessed on 30 October 2012).
- Garcia, P.; Martinez, B.; Obeso, J. M.; Lavigne, R.; Lurz, R.; Rodriguez, A. Functional genomic analysis of two Staphylococcus aureus phages isolated from the dairy environment. Appl. Environ. Microbiol. 2009, 75, 7663–7673. [Google Scholar] [CrossRef]
- Kim, M.S.; Myung, H. Complete Genome of Staphylococcus aureus Phage SA11. J. Virol. 2012, 86, 10232. [Google Scholar] [CrossRef]
- Ackermann, H.W. Tailed bacteriophages: the order caudovirales. Adv. Virus Res. 1998, 51, 135–201. [Google Scholar] [CrossRef]
- Son, J.S.; Lee, S.J.; Jun, S.Y.; Yoon, S.J.; Kang, S.H.; Paik, H.R.; Kang, J.O.; Choi, Y.J. Antibacterial and biofilm removal activity of a podoviridae Staphylococcus aureus bacteriophage SAP-2 and a derived recombinant cell-wall-degrading enzyme. Appl. Microbiol. Biotechnol. 2010, 86, 1439–1449. [Google Scholar] [CrossRef]
- O'Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of Staphylococcal Phage K: a New Lineage of Myoviridae Infecting Gram-Positive Bacteria with a Low G+C Content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; Brucker, W.; Kumar, V.; Kandasamy, J.; Keenan, L.; Bardarov, S.; Kriakov, J.; Lawrence, J.G.; Jacobs, W.R., Jr.; Hendrix, R.W.; Hatfull, G.F. Origins of highly mosaic mycobacteriophage genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; Dubow, M.; Gros, P.; Pelletier, J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 2006, 188, 1184–1187. [Google Scholar] [CrossRef]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef]
- Bae, T.; Baba, T.; Hiramatsu, K.; Schneewind, O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006, 62, 1035–1047. [Google Scholar] [CrossRef]
- Vybiral, D.; Takac, M.; Loessner, M.; Witte, A.; von Ahsen, U.; Blasi, U. Complete nucleotide sequence and molecular characterization of two lytic Staphylococcus aureus phages: 44AHJD and P68. FEMS Microbiol. Lett. 2003, 219, 275–283. [Google Scholar] [CrossRef]
- Lobocka, M.; Hejnowicz, M.S.; Dabrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dabrowska, B.; Kwiatek, M.; Parasion, S.; Gawor, J.; Kosowska, H.; Glowacka, A. Genomics of staphylococcal Twort-like phages--potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [CrossRef]
- Krisch, H.M.; Comeau, A.M. The immense journey of bacteriophage T4--from d'Herelle to Delbruck and then to Darwin and beyond. Res. Microbiol. 2008, 159, 314–324. [Google Scholar] [CrossRef]
- Holden, M.T.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; Barron, A.; Bason, N.; Bentley, S.D.; Chillingworth, C.; Chillingworth, T.; Churcher, C.; Clark, L.; Corton, C.; Cronin, A.; Doggett, J.; Dowd, L.; Feltwell, T.; Hance, Z.; Harris, B.; Hauser, H.; Holroyd, S.; Jagels, K.; James, K. D.; Lennard, N.; Line, A.; Mayes, R.; Moule, S.; Mungall, K.; Ormond, D.; Quail, M.A.; Rabbinowitsch, E.; Rutherford, K.; Sanders, M.; Sharp, S.; Simmonds, M.; Stevens, K.; Whitehead, S.; Barrell, B.G.; Spratt, B.G.; Parkhill, J. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 2004, 101, 9786–9791. [Google Scholar]
- Lindsay, J.A.; Holden, M.T. Staphylococcus aureus: superbug, super genome? Trends Microbiol. 2004, 12, 378–385. [Google Scholar] [CrossRef]
- Suzuki, H.; Lefebure, T.; Bitar, P.P.; Stanhope, M.J. Comparative genomic analysis of the genus Staphylococcus including Staphylococcus aureus and its newly described sister species Staphylococcus simiae. BMC Genomics 2012, 13, 38. [Google Scholar] [CrossRef]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef]
- Coleman, D.C.; Sullivan, D.J.; Russell, R.J.; Arbuthnott, J.P.; Carey, B.F.; Pomeroy, H.M. Staphylococcus aureus bacteriophages mediating the simultaneous lysogenic conversion of beta-lysin, staphylokinase and enterotoxin A: molecular mechanism of triple conversion. J. Gen. Microbiol. 1989, 135, 1679–1697. [Google Scholar]
- van Wamel, W.J.; Rooijakkers, S.H.; Ruyken, M.; van Kessel, K.P.; van Strijp, J.A. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 2006, 188, 1310–1315. [Google Scholar] [CrossRef]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef]
- Zou, D.; Kaneko, J.; Narita, S.; Kamio, Y. Prophage, phiPV83-pro, carrying panton-valentine leukocidin genes, on the Staphylococcus aureus P83 chromosome: comparative analysis of the genome structures of phiPV83-pro, phiPVL, phi11, and other phages. Biosci. Biotechnol. Biochem. 2000, 64, 2631–2643. [Google Scholar] [CrossRef]
- Goerke, C.; Pantucek, R.; Holtfreter, S.; Schulte, B.; Zink, M.; Grumann, D.; Broker, B.M.; Doskar, J.; Wolz, C. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 2009, 191, 3462–3468. [Google Scholar]
- Deghorain, M.; Bobay, L.M.; Smeesters, P.R.; Bousbata, S.; Vermeersch, M.; Perez-Morga, D.; Dreze, P.A.; Rocha, E.P.; Touchon, M.; Van Melderen, L. Characterization of novel phages isolated in coagulase-negative Staphylococci reveals evolutionary relationships with S. aureus phages. J. Bacteriol. 2012, 194, 5829–5839. [Google Scholar] [CrossRef]
- Goerke, C.; Koller, J.; Wolz, C. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 171–177. [Google Scholar] [CrossRef]
- Sumby, P.; Waldor, M.K. Transcription of the toxin genes present within the Staphylococcal phage phiSa3ms is intimately linked with the phage's life cycle. J. Bacteriol. 2003, 185, 6841–6851. [Google Scholar] [CrossRef]
- Novick, R.P.; Christie, G.E.; Penades, J.R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010, 8, 541–551. [Google Scholar] [CrossRef]
- Lindsay, J.A.; Ruzin, A.; Ross, H.F.; Kurepina, N.; Novick, R.P. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 1998, 29, 527–543. [Google Scholar] [CrossRef]
- Tormo-Mas, M.A.; Mir, I.; Shrestha, A.; Tallent, S.M.; Campoy, S.; Lasa, I.; Barbe, J.; Novick, R.P.; Christie, G.E.; Penades, J.R. Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature 2010, 465, 779–782. [Google Scholar]
- Goerke, C.; Wolz, C. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int. J. Med. Microbiol. 2010, 300, 520–525. [Google Scholar] [CrossRef]
- Goerke, C.; Wolz, C. Regulatory and genomic plasticity of Staphylococcus aureus during persistent colonization and infection. Int. J. Med. Microbiol. 2004, 294, 195–202. [Google Scholar] [CrossRef]
- McAdam, P.R.; Holmes, A.; Templeton, K.E.; Fitzgerald, J.R. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One 2011, 6, e24301. [Google Scholar]
- Takeuchi, F.; Watanabe, S.; Baba, T.; Yuzawa, H.; Ito, T.; Morimoto, Y.; Kuroda, M.; Cui, L.; Takahashi, M.; Ankai, A.; Baba, S.; Fukui, S.; Lee, J.C.; Hiramatsu, K. Whole-genome sequencing of staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J. Bacteriol. 2005, 187, 7292–7308. [Google Scholar]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; Deboy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; Dodson, R.J.; Daugherty, S.C.; Madupu, R.; Angiuoli, S.V.; Durkin, A.S.; Haft, D.H.; Vamathevan, J.; Khouri, H.; Utterback, T.; Lee, C.; Dimitrov, G.; Jiang, L.; Qin, H.; Weidman, J.; Tran, K.; Kang, K.; Hance, I.R.; Nelson, K.E.; Fraser, C.M. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar]
- Rosenstein, R.; Nerz, C.; Biswas, L.; Resch, A.; Raddatz, G.; Schuster, S.C.; Gotz, F. Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl. Environ. Microbiol. 2009, 75, 811–822. [Google Scholar] [CrossRef]
- Rosenstein, R.; Gotz, F. Genomic differences between the food-grade Staphylococcus carnosus and pathogenic staphylococcal species. Int. J. Med. Microbiol. 2010, 300, 104–108. [Google Scholar] [CrossRef]
- Lina, B.; Bes, M.; Vandenesch, F.; Greenland, T.; Etienne, J.; Fleurette, J. Role of bacteriophages in genomic variability of related coagulase-negative staphylococci. FEMS Microbiol. Lett. 1993, 109, 273–277. [Google Scholar] [CrossRef]
- Gutierrez, D.; Martinez, B.; Rodriguez, A.; Garcia, P. Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genomics 2012, 13, 228. [Google Scholar] [CrossRef] [Green Version]
- Aswani, V.; Tremblay, D.M.; Moineau, S.; Shukla, S.K. Staphylococcus epidermidis bacteriophages from the anterior nares of humans. Appl. Environ. Microbiol. 2011, 77, 7853–7855. [Google Scholar] [CrossRef]
- Boussard, P.; Pithsy, A.; Devleeschouwer, M.J.; Dony, J. Phage typing of coagulase-negative staphylococci. J. Clin. Pharm. Ther. 1992, 17, 165–168. [Google Scholar]
- Barcs, I.; Herendi, A.; Lipcsey, A.; Bognar, C.; Hashimoto, H. Phage pattern and antibiotic resistance pattern of coagulase-negative staphylococci obtained from immunocompromised patients. Microbiol. Immunol. 1992, 36, 947–959. [Google Scholar]
- Bes, M.; Ackermann, H.W.; Brun, Y.; Fleurette, J. Morphology of Staphylococcus saprophyticus bacteriophages. Res. Virol. 1990, 141, 625–635. [Google Scholar] [CrossRef]
- Bes, M. Characterization of thirteen Staphylococcus epidermidis and S. saprophyticus bacteriophages. Res. Virol. 1994, 145, 111–121. [Google Scholar] [CrossRef]
- Gutierrez, D.; Martinez, B.; Rodriguez, A.; Garcia, P. Isolation and characterization of bacteriophages infecting Staphylococcus epidermidis. Curr. Microbiol. 2010, 61, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Daniel, A.; Bonnen, P.E.; Fischetti, V.A. First complete genome sequence of two Staphylococcus epidermidis bacteriophages. J. Bacteriol. 2007, 189, 2086–2100. [Google Scholar] [CrossRef]
- Madhusoodanan, J.; Seo, K.S.; Remortel, B.; Park, J.Y.; Hwang, S.Y.; Fox, L.K.; Park, Y.H.; Deobald, C.F.; Wang, D.; Liu, S.; Daugherty, S.C.; Gill, A.L.; Bohach, G.A.; Gill, S.R. An Enterotoxin-Bearing Pathogenicity Island in Staphylococcus epidermidis. J. Bacteriol. 2011, 193, 1854–1862. [Google Scholar]
- Ackermann, H.W.; DuBow, M.S.; Jarvis, A.W.; Jones, L.A.; Krylov, V.N.; Maniloff, J.; Rocourt, J.; Safferman, R.S.; Schneider, J.; Seldin, L.; et al. The species concept and its application to tailed phages. Arch. Virol. 1992, 124, 69–82. [Google Scholar] [CrossRef]
- Lee, J.S.; Stewart, P.R. The virion proteins and ultrastructure of Staphylococcus aureus bacteriophages. J. Gen. Virol. 1985, 66, 2017–2027. [Google Scholar] [CrossRef]
- Stewart, P.R.; Waldron, H.G.; Lee, J.S.; Matthews, P.R. Molecular relationships among serogroup B bacteriophages of Staphylococcus aureus. J. Virol. 1985, 55, 111–116. [Google Scholar]
- Pantucek, R.; Doskar, J.; Ruzickova, V.; Kasparek, P.; Oracova, E.; Kvardova, V.; Rosypal, S. Identification of bacteriophage types and their carriage in Staphylococcus aureus. Arch. Virol. 2004, 149, 1689–1703. [Google Scholar] [CrossRef]
- Brussow, H.; Desiere, F. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 2001, 39, 213–222. [Google Scholar] [CrossRef]
- Le Marrec, C.; van Sinderen, D.; Walsh, L.; Stanley, E.; Vlegels, E.; Moineau, S.; Heinze, P.; Fitzgerald, G.; Fayard, B. Two groups of bacteriophages infecting Streptococcus thermophilus can be distinguished on the basis of mode of packaging and genetic determinants for major structural proteins. Appl. Environ. Microbiol. 1997, 63, 3246–3253. [Google Scholar]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Chibani-Chennoufi, S.; Dillmann, M.L.; Marvin-Guy, L.; Rami-Shojaei, S.; Brussow, H. Lactobacillus plantarum bacteriophage LP65: a new member of the SPO1-like genus of the family Myoviridae. J. Bacteriol. 2004, 186, 7069–7083. [Google Scholar] [CrossRef]
- Grossi, P.A. Early appropriate therapy of Gram-positive bloodstream infections: the conservative use of new drugs. Int. J. Antimicrob. Agents 2009, 34 Suppl. 4, S31–34. [Google Scholar] [CrossRef]
- Kahankova, J.; Pantucek, R.; Goerke, C.; Ruzickova, V.; Holochova, P.; Doskar, J. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ. Microbiol. 2010, 12, 2527–2538. [Google Scholar] [CrossRef]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Toussaint, A.; Leplae, R. A modular view of the bacteriophage genomic space: identification of host and lifestyle marker modules. Res. Microbiol. 2011, 162, 737–746. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef]
- Ma, X.X.; Ito, T.; Kondo, Y.; Cho, M.; Yoshizawa, Y.; Kaneko, J.; Katai, A.; Higashiide, M.; Li, S.; Hiramatsu, K. Two different Panton-Valentine leukocidin phage lineages predominate in Japan. J. Clin. Microbiol. 2008, 46, 3246–3258. [Google Scholar] [CrossRef]
- Ma, X.X.; Ito, T.; Chongtrakool, P.; Hiramatsu, K. Predominance of clones carrying Panton-Valentine leukocidin genes among methicillin-resistant Staphylococcus aureus strains isolated in Japanese hospitals from 1979 to 1985. J. Clin. Microbiol. 2006, 44, 4515–4527. [Google Scholar] [CrossRef]
- Narita, S.; Kaneko, J.; Chiba, J.; Piemont, Y.; Jarraud, S.; Etienne, J.; Kamio, Y. Phage conversion of Panton-Valentine leukocidin in Staphylococcus aureus: molecular analysis of a PVL-converting phage, phiSLT. Gene 2001, 268, 195–206. [Google Scholar] [CrossRef]
- Iandolo, J.J.; Worrell, V.; Groicher, K.H.; Qian, Y.; Tian, R.; Kenton, S.; Dorman, A.; Ji, H.; Lin, S.; Loh, P.; Qi, S.; Zhu, H.; Roe, B. A. Comparative analysis of the genomes of the temperate bacteriophages phi 11, phi 12 and phi 13 of Staphylococcus aureus 8325. Gene 2002, 289, 109–118. [Google Scholar] [CrossRef]
- Zhang, M.; Ito, T.; Li, S.; Jin, J.; Takeuchi, F.; Lauderdale, T.L.; Higashide, M.; Hiramatsu, K. Identification of the third type of PVL phage in ST59 methicillin-resistant Staphylococcus aureus (MRSA) strains. FEMS Microbiol. Lett. 2011, 323, 20–28. [Google Scholar] [CrossRef]
- Belcaid, M.; Bergeron, A.; Poisson, G. Mosaic graphs and comparative genomics in phage communities. J. Comput. Biol. 2010, 17, 1315–1326. [Google Scholar] [CrossRef]
- Susskind, M.M.; Botstein, D. Molecular genetics of bacteriophage P22. Microbiol. Rev. 1978, 42, 385–413. [Google Scholar]
- Clark, A.J.; Inwood, W.; Cloutier, T.; Dhillon, T.S. Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J. Mol. Biol. 2001, 311, 657–679. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophages: evolution of the majority. Theor. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Pedulla, M.L.; Jacobs-Sera, D.; Cichon, P.M.; Foley, A.; Ford, M.E.; Gonda, R.M.; Houtz, J.M.; Hryckowian, A.J.; Kelchner, V.A.; Namburi, S.; Pajcini, K.V.; Popovich, M.G.; Schleicher, D.T.; Simanek, B.Z.; Smith, A.L.; Zdanowicz, G.M.; Kumar, V.; Peebles, C.L.; Jacobs, W.R., Jr.; Lawrence, J.G.; Hendrix, R.W. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2006, 2, e92. [Google Scholar] [CrossRef] [Green Version]
- Glazko, G.; Makarenkov, V.; Liu, J.; Mushegian, A. Evolutionary history of bacteriophages with double-stranded DNA genomes. Biol. Direct. 2007, 2, 36. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef]
- Mann, N.H. The potential of phages to prevent MRSA infections. Res. Microbiol. 2008, 159, 400–405. [Google Scholar] [CrossRef]
- Lu, T.K.; Koeris, M.S. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 2011, 14, 524–531. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Yasuda, M.; Nishikawa, H.; Kuroda, M.; Ujihara, T.; Shuin, T.; Shen, Y.; Jin, Z.; Fujimoto, S.; Nasimuzzaman, M.D.; Wakiguchi, H.; Sugihara, S.; Sugiura, T.; Koda, S.; Muraoka, A.; Imai, S. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J. Infect. Dis. 2003, 187, 613–624. [Google Scholar] [CrossRef]
- Kwiatek, M.; Parasion, S.; Mizak, L.; Gryko, R.; Bartoszcze, M.; Kocik, J. Characterization of a bacteriophage, isolated from a cow with mastitis, that is lytic against Staphylococcus aureus strains. Arch. Virol. 2012, 157, 225–234. [Google Scholar] [CrossRef]
- Hagens, S.; Loessner, M.J. Bacteriophage for biocontrol of foodborne pathogens: calculations and considerations. Curr. Pharm. Biotechnol. 2010, 11, 58–68. [Google Scholar] [CrossRef]
- Bueno, E.; Garcia, P.; Martinez, B.; Rodriguez, A. Phage inactivation of Staphylococcus aureus in fresh and hard-type cheeses. Int. J. Food Microbiol. 2012, 158, 23–27. [Google Scholar] [CrossRef]
- Garcia, P.; Madera, C.; Martinez, B.; Rodriguez, A.; Evaristo Suarez, J. Prevalence of bacteriophages infecting Staphylococcus aureus in dairy samples and their potential as biocontrol agents. J. Dairy Sci. 2009, 92, 3019–3026. [Google Scholar] [CrossRef]
- Garcia, P.; Martinez, B.; Obeso, J.M.; Rodriguez, A. Bacteriophages and their application in food safety. Lett. Appl. Microbiol. 2008, 47, 479–485. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef]
- O'Flaherty, S.; Coffey, A.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. The recombinant phage lysin LysK has a broad spectrum of lytic activity against clinically relevant staphylococci, including methicillin-resistant Staphylococcus aureus. J. Bacteriol. 2005, 187, 7161–7164. [Google Scholar] [CrossRef]
- O'Flaherty, S.; Ross, R.P.; Meaney, W.; Fitzgerald, G.F.; Elbreki, M.F.; Coffey, A. Potential of the polyvalent anti-Staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl. Environ. Microbiol. 2005, 71, 1836–1842. [Google Scholar] [CrossRef]
- Hsieh, S.E.; Lo, H.H.; Chen, S.T.; Lee, M.C.; Tseng, Y.H. Wide host range and strong lytic activity of Staphylococcus aureus lytic phage Stau2. Appl. Environ. Microbiol. 2011, 77, 756–761. [Google Scholar] [CrossRef]
- O'Flaherty, S.; Ross, R.P.; Flynn, J.; Meaney, W.J.; Fitzgerald, G.F.; Coffey, A. Isolation and characterization of two anti-staphylococcal bacteriophages specific for pathogenic Staphylococcus aureus associated with bovine infections. Lett. Appl. Microbiol. 2005, 41, 482–486. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; Lavigne, R.; Volckaert, G.; Mattheus, W.; Verween, G.; De Corte, P.; Rose, T.; Jennes, S.; Zizi, M.; De Vos, D.; Vaneechoutte, M. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS One 2009, 4, e4944. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deghorain, M.; Van Melderen, L. The Staphylococci Phages Family: An Overview. Viruses 2012, 4, 3316-3335. https://doi.org/10.3390/v4123316
Deghorain M, Van Melderen L. The Staphylococci Phages Family: An Overview. Viruses. 2012; 4(12):3316-3335. https://doi.org/10.3390/v4123316
Chicago/Turabian StyleDeghorain, Marie, and Laurence Van Melderen. 2012. "The Staphylococci Phages Family: An Overview" Viruses 4, no. 12: 3316-3335. https://doi.org/10.3390/v4123316
APA StyleDeghorain, M., & Van Melderen, L. (2012). The Staphylococci Phages Family: An Overview. Viruses, 4(12), 3316-3335. https://doi.org/10.3390/v4123316