Host Cell Factors in Filovirus Entry: Novel Players, New Insights


Abstract
:1. Introduction
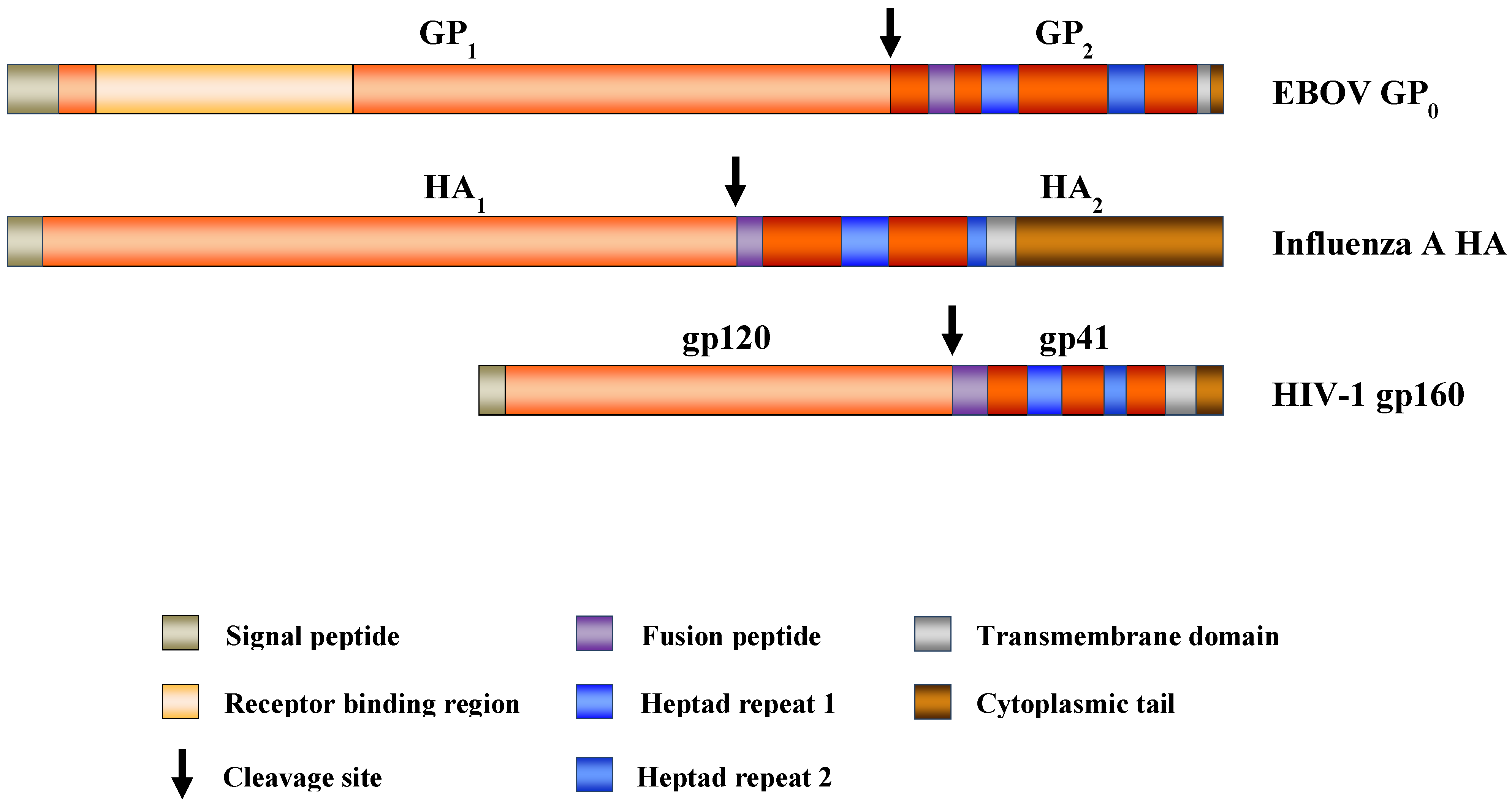
2. The Filoviral Glycoprotein: Structure and Function
2.1. Biosynthesis and Posttranslational Modification of the Filovirus Glycoprotein
2.2. The Filovirus Glycoprotein Is a Class I Membrane Fusion Protein

2.3. Filovirus Fusion with Host Cell Membranes
3. Cell and Organ Tropism of Filoviruses
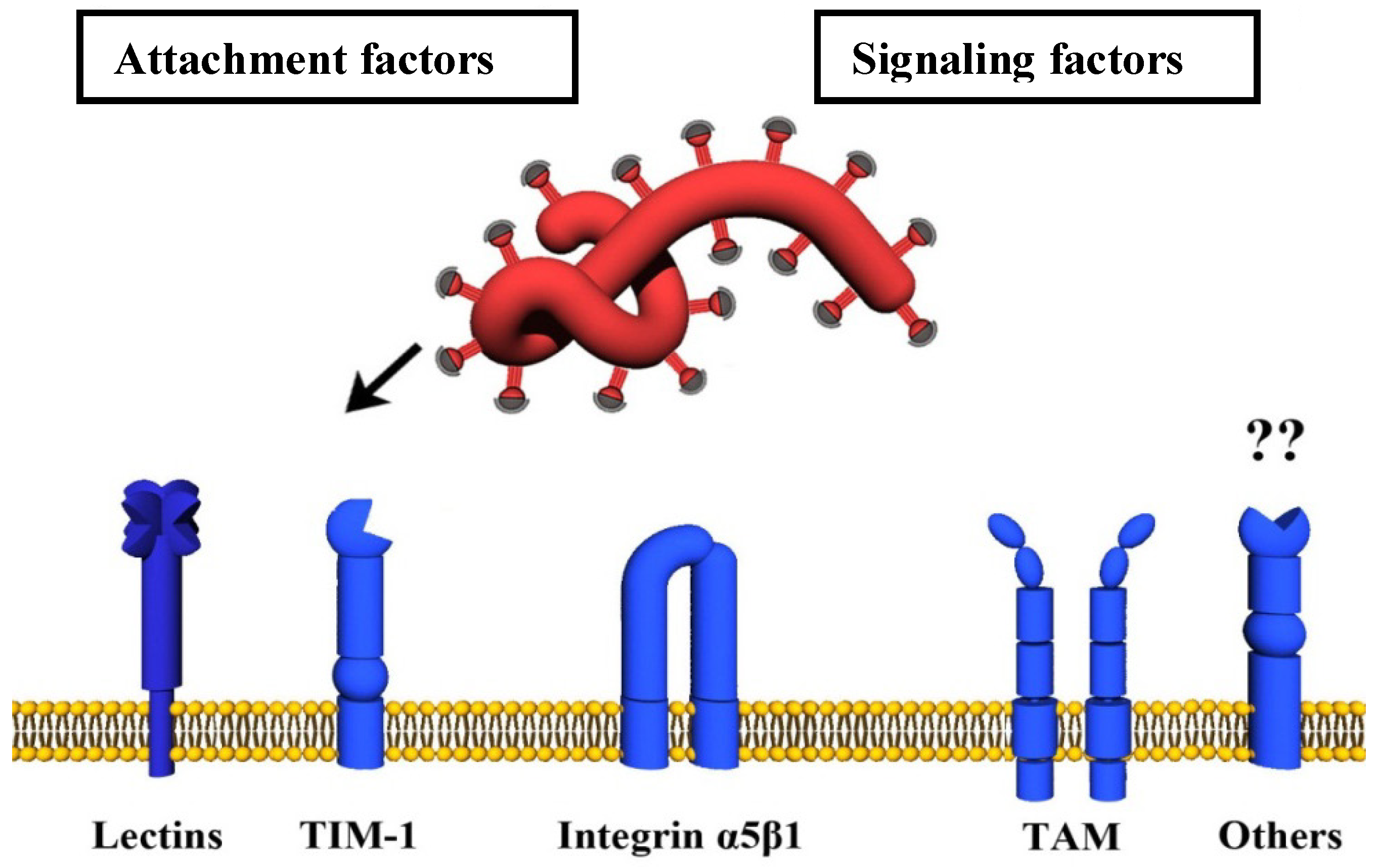
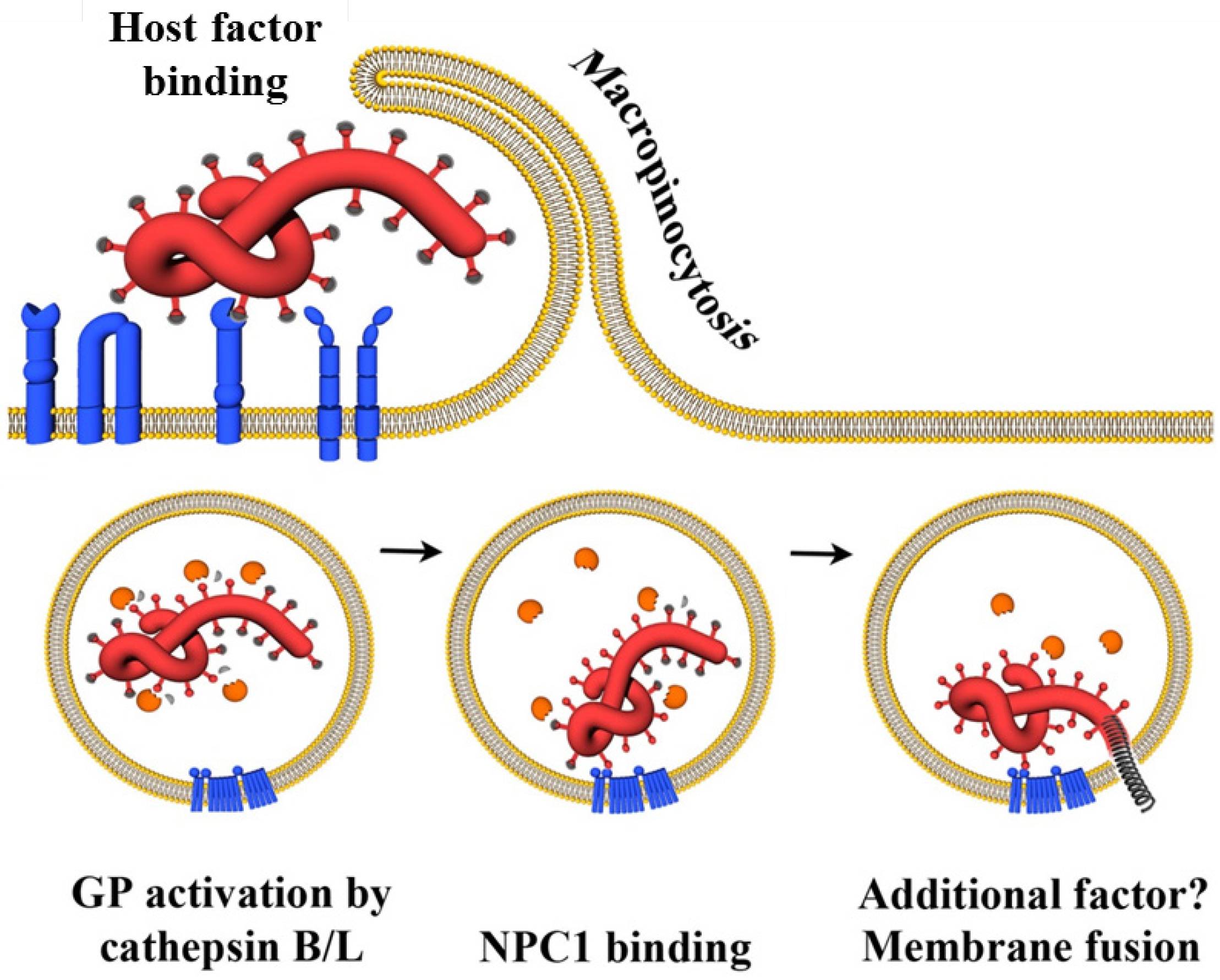
4. Host Cell Factors Promoting Infectious Filovirus Entry

4.1. Attachment Factors
4.1.1. Carbohydrate-Binding Host Cell Factors (Lectins)

{kind=link}
{kind=link}
{kind=link}
| GP-binding lectins | Tissue distribution | Reference |
|---|---|---|
| ASGPR-1 |
| [86] |
| hMGL |
| [41] |
| DC-SIGN |
| [85,85,88,90,101,102,103] |
| DC-SIGNR |
| [85] |
| LSECtin |
| [34,87,102] |
4.1.2. TIM-1
| Host factor | Expression | Analysis in cell lines | Reference |
|---|---|---|---|
| TIM-1 |
|
| [105,109,110] |
| Axl |
|
| [114,115,116,117,118,119,120] |
| Integrin αV β1 |
|
| [121,122,123,124] |
| Cathepsin B and L |
|
| [24,56,57,125,126,127,128] |
| NPC1 |
|
| [20,129,130] |
4.2. Signaling Factors
4.2.1. TAM Family of Tyrosine Kinase Receptors
4.2.2. α5β1-Integrin
4.3. Endo-/Lysosomal Host Cell Factors
4.3.1. Cathepsins B and L

4.3.2. NPC1
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Mahanty, S.; Bray, M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 2004, 4, 487–498. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Jahrling, P.B.; Kawaoka, Y.; Netesov, S.V.; Nichol, S.T.; Peters, C.J.; Volchkov, V.E.; et al. Family Filoviridae. In Virus Taxonomy—Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier/Academic Press: London, UK, 2001; pp. 665–671. [Google Scholar]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef]
- Hutchinson, K.L.; Rollin, P.E. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J. Infect. Dis. 2007, 196, S357–S363. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Dodd, L.E.; Wahl-Jensen, V.; Radoshitzky, S.R.; Bavari, S.; Jahrling, P.B. Evaluation of perceived threat differences posed by filovirus variants. Biosecur. Bioterror. 2011, 9, 361–371. [Google Scholar] [CrossRef]
- Barrette, R.W.; Metwally, S.A.; Rowland, J.M.; Xu, L.; Zaki, S.R.; Nichol, S.T.; Rollin, P.E.; Towner, J.S.; Shieh, W.J.; Batten, B.; et al. Discovery of swine as a host for the Reston ebolavirus. Science 2009, 325, 204–206. [Google Scholar]
- Miranda, M.E.; Ksiazek, T.G.; Retuya, T.J.; Khan, A.S.; Sanchez, A.; Fulhorst, C.F.; Rollin, P.E.; Calaor, A.B.; Manalo, D.L.; Roces, M.C.; et al. Epidemiology of Ebola (subtype Reston) virus in the Philippines, 1996. J. Infect. Dis. 1999, 179, S115–S119. [Google Scholar]
- Rollin, P.E.; Williams, R.J.; Bressler, D.S.; Pearson, S.; Cottingham, M.; Pucak, G.; Sanchez, A.; Trappier, S.G.; Peters, R.L.; Greer, P.W.; et al. Ebola (subtype Reston) virus among quarantined nonhuman primates recently imported from the Philippines to the United States. J. Infect. Dis. 1999, 179, S108–S114. [Google Scholar]
- Jahrling, P.B.; Geisbert, T.W.; Jaax, N.K.; Hanes, M.A.; Ksiazek, T.G.; Peters, C.J. Experimental infection of cynomolgus macaques with Ebola-Reston filoviruses from the 1989–1990 U.S. epizootic. Arch. Virol. Suppl. 1996, 11, 115–134. [Google Scholar]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar]
- Pourrut, X.; Souris, M.; Towner, J.S.; Rollin, P.E.; Nichol, S.T.; Gonzalez, J.P.; Leroy, E. Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect. Dis. 2009, 9, 159. [Google Scholar] [CrossRef]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Niezgoda, M.; Franka, R.; Agwanda, B.; Markotter, W.; Breiman, R.F.; Shieh, W.J.; Zaki, S.R.; Rupprecht, C.E. Marburg virus in fruit bat, Kenya. Emerg. Infect. Dis. 2010, 16, 352–354. [Google Scholar] [CrossRef]
- Taniguchi, S.; Watanabe, S.; Masangkay, J.S.; Omatsu, T.; Ikegami, T.; Alviola, P.; Ueda, N.; Iha, K.; Fujii, H.; Ishii, Y.; et al. Reston Ebolavirus antibodies in bats, the Philippines. Emerg. Infect. Dis. 2011, 17, 1559–1560. [Google Scholar]
- Negredo, A.; Palacios, G.; Vazquez-Moron, S.; Gonzalez, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; de la Cruz, M.M.; et al. Discovery of an ebolavirus-like filovirus in europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar]
- Leroy, E.M.; Rouquet, P.; Formenty, P.; Souquiere, S.; Kilbourne, A.; Froment, J.M.; Bermejo, M.; Smit, S.; Karesh, W.; Swanepoel, R.; et al. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 2004, 303, 387–390. [Google Scholar] [CrossRef]
- Bermejo, M.; Rodriguez-Teijeiro, J.D.; Illera, G.; Barroso, A.; Vila, C.; Walsh, P.D. Ebola outbreak killed 5000 gorillas. Science 2006, 314, 1564. [Google Scholar] [CrossRef]
- Sayama, Y.; Demetria, C.; Saito, M.; Azul, R.R.; Taniguchi, S.; Fukushi, S.; Yoshikawa, T.; Iizuka, I.; Mizutani, T.; Kurane, I.; et al. A seroepidemiologic study of Reston ebolavirus in swine in the Philippines. BMC Vet. Res. 2012, 8, 82. [Google Scholar] [CrossRef]
- Miller, E.H.; Harrison, J.S.; Radoshitzky, S.R.; Higgins, C.D.; Chi, X.; Dong, L.; Kuhn, J.H.; Bavari, S.; Lai, J.R.; Chandran, K. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 2011, 286, 15854–15861. [Google Scholar]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008, 4, e1000141. [Google Scholar] [CrossRef]
- Barrientos, L.G.; O'Keefe, B.R.; Bray, M.; Sanchez, A.; Gronenborn, A.M.; Boyd, M.R. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antivir. Res. 2003, 58, 47–56. [Google Scholar]
- Basu, A.; Li, B.; Mills, D.M.; Panchal, R.G.; Cardinale, S.C.; Butler, M.M.; Peet, N.P.; Majgier-Baranowska, H.; Williams, J.D.; Patel, I.; et al. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 2011, 85, 3106–3119. [Google Scholar] [CrossRef]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar] [CrossRef]
- Groseth, A.; Marzi, A.; Hoenen, T.; Herwig, A.; Gardner, D.; Becker, S.; Ebihara, H.; Feldmann, H. The Ebola virus glycoprotein contributes to but is not sufficient for virulence in vivo. PLoS Pathog. 2012, 8, e1002847. [Google Scholar] [CrossRef]
- Takada, A.; Robison, C.; Goto, H.; Sanchez, A.; Murti, K.G.; Whitt, M.A.; Kawaoka, Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 14764–14769. [Google Scholar]
- Wool-Lewis, R.J.; Bates, P. Characterization of Ebola virus entry by using pseudotyped viruses: Identification of receptor-deficient cell lines. J. Virol. 1998, 72, 3155–3160. [Google Scholar]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef]
- Feldmann, H.; Volchkov, V.E.; Volchkova, V.A.; Klenk, H.D. The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 1999, 15, 159–169. [Google Scholar]
- Hoenen, T.; Groseth, A.; Falzarano, D.; Feldmann, H. Ebola virus: Unravelling pathogenesis to combat a deadly disease. Trends Mol. Med. 2006, 12, 206–215. [Google Scholar] [CrossRef]
- Takada, A.; Kawaoka, Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 2001, 9, 506–511. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 5762–5767. [Google Scholar] [CrossRef]
- Sanchez, A.; Yang, Z.Y.; Xu, L.; Nabel, G.J.; Crews, T.; Peters, C.J. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 1998, 72, 6442–6447. [Google Scholar]
- Powlesland, A.S.; Fisch, T.; Taylor, M.E.; Smith, D.F.; Tissot, B.; Dell, A.; Pöhlmann, S.; Drickamer, K. A novel mechanism for LSECtin binding to Ebola virus surface glycoprotein through truncated glycans. J. Biol. Chem. 2008, 283, 593–602. [Google Scholar]
- Ritchie, G.; Harvey, D.J.; Stroeher, U.; Feldmann, F.; Feldmann, H.; Wahl-Jensen, V.; Royle, L.; Dwek, R.A.; Rudd, P.M. Identification of N-glycans from Ebola virus glycoproteins by matrix-assisted laser desorption/ionisation time-of-flight and negative ion electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 571–585. [Google Scholar] [CrossRef]
- Manicassamy, B.; Wang, J.; Rumschlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.; Rong, L. Characterization of Marburg virus glycoprotein in viral entry. Virology 2007, 358, 79–88. [Google Scholar] [CrossRef]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 3602–3607. [Google Scholar]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent modifications of the ebola virus glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar] [CrossRef]
- Simmons, G.; Wool-Lewis, R.J.; Baribaud, F.; Netter, R.C.; Bates, P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002, 76, 2518–2528. [Google Scholar] [CrossRef]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004, 78, 2943–2947. [Google Scholar] [CrossRef]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 2662–2667. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Carfi, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomai. Mol. Cell 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Radoshitzky, S.R.; Guth, A.C.; Warfield, K.L.; Li, W.; Vincent, M.J.; Towner, J.S.; Nichol, S.T.; Bavari, S.; Choe, H.; et al. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J. Biol. Chem. 2006, 281, 15951–15958. [Google Scholar]
- Neumann, G.; Geisbert, T.W.; Ebihara, H.; Geisbert, J.B.; Daddario-DiCaprio, K.M.; Feldmann, H.; Kawaoka, Y. Proteolytic processing of the Ebola virus glycoprotein is not critical for Ebola virus replication in nonhuman primates. J. Virol. 2007, 81, 2995–2998. [Google Scholar]
- Neumann, G.; Feldmann, H.; Watanabe, S.; Lukashevich, I.; Kawaoka, Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 2002, 76, 406–410. [Google Scholar] [CrossRef]
- Jaskierny, A.J.; Panahi, A.; Feig, M. Effect of flanking residues on the conformational sampling of the internal fusion peptide from Ebola virus. Proteins 2011, 79, 1109–1117. [Google Scholar] [CrossRef]
- Watanabe, S.; Takada, A.; Watanabe, T.; Ito, H.; Kida, H.; Kawaoka, Y. Functional importance of the coiled-coil of the Ebola virus glycoprotein. J. Virol. 2000, 74, 10194–10201. [Google Scholar] [CrossRef]
- Usami, K.; Matsuno, K.; Igarashi, M.; Denda-Nagai, K.; Takada, A.; Irimura, T. Involvement of viral envelope GP2 in Ebola virus entry into cells expressing the macrophage galactose-type C-type lectin. Biochem. Biophys. Res. Commun. 2011, 407, 74–78. [Google Scholar] [CrossRef]
- Adam, B.; Lins, L.; Stroobant, V.; Thomas, A.; Brasseur, R. Distribution of hydrophobic residues is crucial for the fusogenic properties of the Ebola virus GP2 fusion peptide. J. Virol. 2004, 78, 2131–2136. [Google Scholar]
- Ito, H.; Watanabe, S.; Sanchez, A.; Whitt, M.A.; Kawaoka, Y. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J. Virol. 1999, 73, 8907–8912. [Google Scholar]
- Ruiz-Arguello, M.B.; Goni, F.M.; Pereira, F.B.; Nieva, J.L. Phosphatidylinositol-dependent membrane fusion induced by a putative fusogenic sequence of Ebola virus. J. Virol. 1998, 72, 1775–1781. [Google Scholar]
- Hofmann, H.; Pöhlmann, S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004, 12, 466–472. [Google Scholar] [CrossRef]
- Manicassamy, B.; Wang, J.; Jiang, H.; Rong, L. Comprehensive analysis of ebola virus GP1 in viral entry. J. Virol. 2005, 79, 4793–4805. [Google Scholar]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Dias, J.M.; Kuehne, A.I.; Abelson, D.M.; Bale, S.; Wong, A.C.; Halfmann, P.; Muhammad, M.A.; Fusco, M.L.; Zak, S.E.; Kang, E.; et al. A shared structural solution for neutralizing ebolaviruses. Nat. Struct. Mol. Biol. 2011, 18, 1424–1427. [Google Scholar] [CrossRef]
- Gregory, S.M.; Harada, E.; Liang, B.; Delos, S.E.; White, J.M.; Tamm, L.K. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11211–11216. [Google Scholar]
- Stroher, U.; West, E.; Bugany, H.; Klenk, H.D.; Schnittler, H.J.; Feldmann, H. Infection and activation of monocytes by Marburg and Ebola viruses. J. Virol. 2001, 75, 11025–11033. [Google Scholar] [CrossRef]
- Gupta, M.; Mahanty, S.; Ahmed, R.; Rollin, P.E. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 2001, 284, 20–25. [Google Scholar] [CrossRef]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jahrling, P.B.; Hanes, M.A.; Zack, P.M. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol. 1992, 106, 137–152. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: Evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar]
- Zaki, S.R.; Goldsmith, C.S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 1999, 235, 97–116. [Google Scholar]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; McRae, M.; Rollin, P.E.; Pulendran, B. Cutting edge: Impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. Ebola and marburg hemorrhagic fever. Clin. Lab. Med. 2010, 30, 161–177. [Google Scholar] [CrossRef]
- Feldmann, H.; Bugany, H.; Mahner, F.; Klenk, H.D.; Drenckhahn, D.; Schnittler, H.J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 1996, 70, 2208–2214. [Google Scholar]
- Bray, M.; Geisbert, T.W. Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005, 37, 1560–1566. [Google Scholar] [CrossRef]
- Schnittler, H.J.; Mahner, F.; Drenckhahn, D.; Klenk, H.D.; Feldmann, H. Replication of Marburg virus in human endothelial cells. A possible mechanism for the development of viral hemorrhagic disease. J. Clin. Invest 1993, 91, 1301–1309. [Google Scholar] [CrossRef]
- Johnson, E.; Jaax, N.; White, J.; Jahrling, P. Lethal experimental infections of rhesus monkeys by aerosolized Ebola virus. Int. J. Exp. Pathol. 1995, 76, 227–236. [Google Scholar]
- Ryabchikova, E.; Strelets, L.; Kolesnikova, L.; Pyankov, O.; Sergeev, A. Respiratory Marburg virus infection in guinea pigs. Arch. Virol. 1996, 141, 2177–2190. [Google Scholar] [CrossRef]
- Ryabchikova, E.; Kolesnikova, L.; Smolina, M.; Tkachev, V.; Pereboeva, L.; Baranova, S.; Grazhdantseva, A.; Rassadkin, Y. Ebola virus infection in guinea pigs: Presumable role of granulomatous inflammation in pathogenesis. Arch. Virol. 1996, 141, 909–921. [Google Scholar] [CrossRef]
- Jaax, N.K.; Davis, K.J.; Geisbert, T.J.; Vogel, P.; Jaax, G.P.; Topper, M.; Jahrling, P.B. Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch. Pathol. Lab. Med. 1996, 120, 140–155. [Google Scholar]
- Davis, K.J.; Anderson, A.O.; Geisbert, T.W.; Steele, K.E.; Geisbert, J.B.; Vogel, P.; Connolly, B.M.; Huggins, J.W.; Jahrling, P.B.; Jaax, N.K. Pathology of experimental Ebola virus infection in African green monkeys. Involvement of fibroblastic reticular cells. Arch. Pathol. Lab. Med. 1997, 121, 805–819. [Google Scholar]
- Chan, S.Y.; Speck, R.F.; Ma, M.C.; Goldsmith, M.A. Distinct mechanisms of entry by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Virol. 2000, 74, 4933–4937. [Google Scholar]
- Ito, H.; Watanabe, S.; Takada, A.; Kawaoka, Y. Ebola virus glycoprotein: Proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 2001, 75, 1576–1580. [Google Scholar] [CrossRef]
- Simmons, G.; Rennekamp, A.J.; Chai, N.; Vandenberghe, L.H.; Riley, J.L.; Bates, P. Folate receptor alpha and caveolae are not required for Ebola virus glycoprotein-mediated viral infection. J. Virol. 2003, 77, 13433–13438. [Google Scholar] [CrossRef]
- Peters, C.J.; Jahrling, P.B.; Ksiazek, T.G.; Johnson, E.D.; Lupton, H.W. Filovirus contamination of cell cultures. Dev. Biol. Stand. 1992, 76, 267–274. [Google Scholar]
- Van der Groen, G. Ebola Virus Hemorraghic Fever; Amsterdam Elsevier/North Holland Biomedical Press: Amsterdam, The Netherlands, 1978. [Google Scholar]
- Yang, Z.; Delgado, R.; Xu, L.; Todd, R.F.; Nabel, E.G.; Sanchez, A.; Nabel, G.J. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 1998, 279, 1034–1037. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muniz, O.; Corbi, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar]
- Becker, S.; Spiess, M.; Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 1995, 76, 393–399. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Moller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef]
- Marzi, A.; Moller, P.; Hanna, S.L.; Harrer, T.; Eisemann, J.; Steinkasserer, A.; Becker, S.; Baribaud, F.; Pöhlmann, S. Analysis of the interaction of Ebola virus glycoprotein with DC-SIGN (dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin) and its homologue DC-SIGNR. J. Infect. Dis. 2007, 196, S237–S246. [Google Scholar] [CrossRef]
- Matsuno, K.; Nakayama, E.; Noyori, O.; Marzi, A.; Ebihara, H.; Irimura, T.; Feldmann, H.; Takada, A. C-type lectins do not act as functional receptors for filovirus entry into cells. Biochem. Biophys. Res. Commun. 2010, 403, 144–148. [Google Scholar] [CrossRef]
- Marzi, A.; Wegele, A.; Pöhlmann, S. Modulation of virion incorporation of Ebolavirus glycoprotein: Effects on attachment, cellular entry and neutralization. Virology 2006, 352, 345–356. [Google Scholar] [CrossRef]
- Matsuno, K.; Kishida, N.; Usami, K.; Igarashi, M.; Yoshida, R.; Nakayama, E.; Shimojima, M.; Feldmann, H.; Irimura, T.; Kawaoka, Y.; et al. Different potential of C-type lectin-mediated entry between Marburg virus strains. J. Virol. 2010, 84, 5140–5147. [Google Scholar]
- Geijtenbeek, T.B.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Adema, G.J.; van, K.Y.; Figdor, C.G. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 2000, 100, 575–585. [Google Scholar] [CrossRef]
- Higashi, N.; Fujioka, K.; Denda-Nagai, K.; Hashimoto, S.; Nagai, S.; Sato, T.; Fujita, Y.; Morikawa, A.; Tsuiji, M.; Miyata-Takeuchi, M.; et al. The macrophage C-type lectin specific for galactose/N-acetylgalactosamine is an endocytic receptor expressed on monocyte-derived immature dendritic cells. J. Biol. Chem. 2002, 277, 20686–20693. [Google Scholar]
- Spiess, M. The asialoglycoprotein receptor: A model for endocytic transport receptors. Biochemistry 1990, 29, 10009–10018. [Google Scholar] [CrossRef]
- Bashirova, A.A.; Geijtenbeek, T.B.; van Duijnhoven, G.C.; van Vliet, S.J.; Eilering, J.B.; Martin, M.P.; Wu, L.; Martin, T.D.; Viebig, N.; Knolle, P.A.; et al. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Exp. Med. 2001, 193, 671–678. [Google Scholar] [CrossRef]
- Pöhlmann, S.; Soilleux, E.J.; Baribaud, F.; Leslie, G.J.; Morris, L.S.; Trowsdale, J.; Lee, B.; Coleman, N.; Doms, R.W. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 2670–2675. [Google Scholar]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den, D.J.; Litjens, M.; Geijtenbeek, T.B. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef]
- Michelow, I.C.; Lear, C.; Scully, C.; Prugar, L.I.; Longley, C.B.; Yantosca, L.M.; Ji, X.; Karpel, M.; Brudner, M.; Takahashi, K.; et al. High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect. Dis. 2011, 203, 175–179. [Google Scholar] [CrossRef]
- Baribaud, F.; Doms, R.W.; Pöhlmann, S. The role of DC-SIGN and DC-SIGNR in HIV and Ebola virus infection: Can potential therapeutics block virus transmission and dissemination? Expert. Opin. Ther. Targets. 2002, 6, 423–431. [Google Scholar] [CrossRef]
- Gramberg, T.; Soilleux, E.; Fisch, T.; Lalor, P.F.; Hofmann, H.; Wheeldon, S.; Cotterill, A.; Wegele, A.; Winkler, T.; Adams, D.H.; et al. Interactions of LSECtin and DC-SIGN/DC-SIGNR with viral ligands: Differential pH dependence, internalization and virion binding. Virology 2008, 373, 189–201. [Google Scholar] [CrossRef]
- Lin, G.; Simmons, G.; Pöhlmann, S.; Baribaud, F.; Ni, H.; Leslie, G.J.; Haggarty, B.S.; Bates, P.; Weissman, D.; Hoxie, J.A.; et al. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 1337–1346. [Google Scholar]
- Kaplan, G.; Totsuka, A.; Thompson, P.; Akatsuka, T.; Moritsugu, Y.; Feinstone, S.M. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996, 15, 4282–4296. [Google Scholar]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 8426–8431. [Google Scholar]
- McIntire, J.J.; Umetsu, S.E.; Akbari, O.; Potter, M.; Kuchroo, V.K.; Barsh, G.S.; Freeman, G.J.; Umetsu, D.T.; DeKruyff, R.H. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat. Immunol. 2001, 2, 1109–1116. [Google Scholar]
- Kobayashi, N.; Karisola, P.; Pena-Cruz, V.; Dorfman, D.M.; Jinushi, M.; Umetsu, S.E.; Butte, M.J.; Nagumo, H.; Chernova, I.; Zhu, B.; et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 2007, 27, 927–940. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Umetsu, S.E.; Lee, W.L.; McIntire, J.J.; Downey, L.; Sanjanwala, B.; Akbari, O.; Berry, G.J.; Nagumo, H.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat. Immunol. 2005, 6, 447–454. [Google Scholar] [CrossRef]
- Ichimura, T.; Asseldonk, E.J.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Invest. 2008, 118, 1657–1668. [Google Scholar] [CrossRef]
- Kuhn, J.H. Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch. Virol. Suppl. 2008, 20, 13–360. [Google Scholar] [CrossRef]
- de Souza, A.J.; Oak, J.S.; Jordanhazy, R.; DeKruyff, R.H.; Fruman, D.A.; Kane, L.P. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. J. Immunol. 2008, 180, 6518–6526. [Google Scholar]
- de Souza, A.J.; Oriss, T.B.; O'malley, K.J.; Ray, A.; Kane, L.P. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17113–17118. [Google Scholar]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B., Jr.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar] [CrossRef]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007, 196, S259–S263. [Google Scholar]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar]
- Crosier, K.E.; Crosier, P.S. New insights into the control of cell growth; the role of the AxI family. Pathology 1997, 29, 131–135. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Berman, A.E.; Kozlova, N.I. Integrins: Structure and functions. Membr. Cell Biol. 2000, 13, 207–244. [Google Scholar]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Schornberg, K.L.; Shoemaker, C.J.; Dube, D.; Abshire, M.Y.; Delos, S.E.; Bouton, A.H.; White, J.M. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8003–8008. [Google Scholar]
- Takada, A.; Watanabe, S.; Ito, H.; Okazaki, K.; Kida, H.; Kawaoka, Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: Implication for virus entry. Virology 2000, 278, 20–26. [Google Scholar] [CrossRef]
- Misasi, J.; Chandran, K.; Yang, J.Y.; Considine, B.; Filone, C.M.; Cote, M.; Sullivan, N.; Fabozzi, G.; Hensley, L.; Cunningham, J. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 2012, 86, 3284–3292. [Google Scholar]
- Kaletsky, R.L.; Simmons, G.; Bates, P. Proteolysis of the Ebola virus glycoproteins enhances virus binding and infectivity. J. Virol. 2007, 81, 13378–13384. [Google Scholar] [CrossRef]
- Sanchez, A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007, 196, S251–S258. [Google Scholar] [CrossRef]
- Martinez, O.; Johnson, J.; Manicassamy, B.; Rong, L.; Olinger, G.G.; Hensley, L.E.; Basler, C.F. Zaire Ebola virus entry into human dendritic cells is insensitive to cathepsin L inhibition. Cell Microbiol. 2010, 12, 148–157. [Google Scholar]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Godowski, P.J.; Mark, M.R.; Chen, J.; Sadick, M.D.; Raab, H.; Hammonds, R.G. Reevaluation of the roles of protein S and Gas6 as ligands for the receptor tyrosine kinase Rse/Tyro 3. Cell 1995, 82, 355–358. [Google Scholar] [CrossRef]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar]
- Hafizi, S.; Dahlback, B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006, 273, 5231–5244. [Google Scholar] [CrossRef]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef]
- Hynes, R.O. Cell adhesion: Old and new questions. Trends Cell Biol. 1999, 9, M33–M37. [Google Scholar] [CrossRef]
- Cary, L.A.; Han, D.C.; Guan, J.L. Integrin-mediated signal transduction pathways. Histol. Histopathol. 1999, 14, 1001–1009. [Google Scholar]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15470–15475. [Google Scholar]
- Mathias, P.; Wickham, T.; Moore, M.; Nemerow, G. Multiple adenovirus serotypes use alpha v integrins for infection. J. Virol. 1994, 68, 6811–6814. [Google Scholar]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Gavrilovskaya, I.N.; Brown, E.J.; Ginsberg, M.H.; Mackow, E.R. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 1999, 73, 3951–3959. [Google Scholar]
- Gavrilovskaya, I.N.; Shepley, M.; Shaw, R.; Ginsberg, M.H.; Mackow, E.R. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7074–7079. [Google Scholar]
- Raymond, T.; Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 1163–1168. [Google Scholar]
- Berinstein, A.; Roivainen, M.; Hovi, T.; Mason, P.W.; Baxt, B. Antibodies to the vitronectin receptor (integrin alpha V beta 3) inhibit binding and infection of foot-and-mouth disease virus to cultured cells. J. Virol. 1995, 69, 2664–2666. [Google Scholar]
- Bergelson, J.M.; Shepley, M.P.; Chan, B.M.; Hemler, M.E.; Finberg, R.W. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science 1992, 255, 1718–1720. [Google Scholar]
- Xing, L.; Huhtala, M.; Pietiainen, V.; Kapyla, J.; Vuorinen, K.; Marjomaki, V.; Heino, J.; Johnson, M.S.; Hyypia, T.; Cheng, R.H. Structural and functional analysis of integrin alpha2I domain interaction with echovirus 1. J. Biol. Chem. 2004, 279, 11632–11638. [Google Scholar]
- Whitton, J.L.; Cornell, C.T.; Feuer, R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 2005, 3, 765–776. [Google Scholar] [CrossRef]
- Graham, K.L.; Fleming, F.E.; Halasz, P.; Hewish, M.J.; Nagesha, H.S.; Holmes, I.H.; Takada, Y.; Coulson, B.S. Rotaviruses interact with alpha4beta7 and alpha4beta1 integrins by binding the same integrin domains as natural ligands. J. Gen. Virol. 2005, 86, 3397–3408. [Google Scholar]
- Maginnis, M.S.; Forrest, J.C.; Kopecky-Bromberg, S.A.; Dickeson, S.K.; Santoro, S.A.; Zutter, M.M.; Nemerow, G.R.; Bergelson, J.M.; Dermody, T.S. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 2006, 80, 2760–2770. [Google Scholar]
- Stewart, P.L.; Nemerow, G.R. Cell integrins: Commonly used receptors for diverse viral pathogens. Trends Microbiol. 2007, 15, 500–507. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Cheresh, D.A. Herpesvirus hijacks an integrin. Nat. Cell Biol. 2002, 4, E69–E71. [Google Scholar] [CrossRef]
- Francica, J.R.; Varela-Rohena, A.; Medvec, A.; Plesa, G.; Riley, J.L.; Bates, P. Steric shielding of surface epitopes and impaired immune recognition induced by the ebola virus glycoprotein. PLoS Pathog. 2010, 6, e1001098. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar]
- Diederich, S.; Sauerhering, L.; Weis, M.; Altmeppen, H.; Schaschke, N.; Reinheckel, T.; Erbar, S.; Maisner, A. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J. Virol. 2012, 86, 3736–3745. [Google Scholar] [CrossRef]
- Popa, A.; Carter, J.R.; Smith, S.E.; Hellman, L.; Fried, M.G.; Dutch, R.E. Residues in the hendra virus fusion protein transmembrane domain are critical for endocytic recycling. J. Virol. 2012, 86, 3014–3026. [Google Scholar]
- White, J.; Kartenbeck, J.; Helenius, A. Membrane fusion activity of influenza virus. EMBO J. 1982, 1, 217–222. [Google Scholar]
- van der Schaar, H.M.; Rust, M.J.; Chen, C.; Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Huotari, J.; Helenius, A. Late-penetrating viruses. Curr. Opin. Virol. 2011, 1, 35–43. [Google Scholar] [CrossRef]
- Mothes, W.; Boerger, A.L.; Narayan, S.; Cunningham, J.M.; Young, J.A. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 2000, 103, 679–689. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Bar, S.; Takada, A.; Kawaoka, Y.; Alizon, M. Detection of cell-cell fusion mediated by Ebola virus glycoproteins. J. Virol. 2006, 80, 2815–2822. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Lysosomal cysteine proteases: More than scavengers. Biochim. Biophys. Acta 2000, 1477, 98–111. [Google Scholar]
- Lutgens, S.P.; Cleutjens, K.B.; Daemen, M.J.; Heeneman, S. Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 2007, 21, 3029–3041. [Google Scholar] [CrossRef]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Invest. 2010, 120, 3421–3431. [Google Scholar] [CrossRef]
- White, J.M.; Schornberg, K.L. A new player in the puzzle of filovirus entry. Nat. Rev. Microbiol. 2012, 10, 317–322. [Google Scholar]
- Dube, D.; Brecher, M.B.; Delos, S.E.; Rose, S.C.; Park, E.W.; Schornberg, K.L.; Kuhn, J.H.; White, J.M. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): Sequence and residues critical for host cell binding. J. Virol. 2009, 83, 2883–2891. [Google Scholar] [CrossRef]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef]
- Bale, S.; Liu, T.; Li, S.; Wang, Y.; Abelson, D.; Fusco, M.; Woods, V.L., Jr.; Saphire, E.O. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl. Trop. Dis. 2011, 5, e1395. [Google Scholar]
- Hood, C.L.; Abraham, J.; Boyington, J.C.; Leung, K.; Kwong, P.D.; Nabel, G.J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: Implications for viral entry and immunogenicity. J. Virol. 2010, 84, 2972–2982. [Google Scholar]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef]
- Gnirss, K.; Kühl, A.; Karsten, C.; Glowacka, I.; Bertram, S.; Kaup, F.; Hofmann, H.; Pohlmann, S. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology 2012, 424, 3–10. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11876–11881. [Google Scholar]
- Roth, W.; Deussing, J.; Botchkarev, V.A.; Pauly-Evers, M.; Saftig, P.; Hafner, A.; Schmidt, P.; Schmahl, W.; Scherer, J.; Anton-Lamprecht, I.; et al. Cathepsin L deficiency as molecular defect of furless: Hyperproliferation of keratinocytes and pertubation of hair follicle cycling. FASEB J. 2000, 14, 2075–2086. [Google Scholar] [CrossRef]
- Benavides, F.; Starost, M.F.; Flores, M.; Gimenez-Conti, I.B.; Guenet, J.L.; Conti, C.J. Impaired hair follicle morphogenesis and cycling with abnormal epidermal differentiation in nackt mice, a cathepsin L-deficient mutation. Am. J. Pathol. 2002, 161, 693–703. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 2000, 106, 1127–1137. [Google Scholar] [CrossRef]
- Wong, A.C.; Sandesara, R.G.; Mulherkar, N.; Whelan, S.P.; Chandran, K. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J. Virol. 2010, 84, 163–175. [Google Scholar] [CrossRef]
- Haines, K.M.; Vande Burgt, N.H.; Francica, J.R.; Kaletsky, R.L.; Bates, P. Chinese hamster ovary cell lines selected for resistance to ebolavirus glycoprotein mediated infection are defective for NPC1 expression. Virology 2012, 432, 20–28. [Google Scholar] [CrossRef]
- Loftus, S.K.; Morris, J.A.; Carstea, E.D.; Gu, J.Z.; Cummings, C.; Brown, A.; Ellison, J.; Ohno, K.; Rosenfeld, M.A.; Tagle, D.A.; et al. Murine model of Niemann-Pick C disease: Mutation in a cholesterol homeostasis gene. Science 1997, 277, 232–235. [Google Scholar] [CrossRef]
- Garver, W.S.; Xie, C.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Niemann-Pick C1 expression is not regulated by the amount of cholesterol flowing through cells in the mouse. J. Lipid. Res. 2005, 46, 1745–1754. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef]
- Karten, B.; Peake, K.B.; Vance, J.E. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim. Biophys. Acta 2009, 1791, 659–670. [Google Scholar]
- Rosenbaum, A.I.; Maxfield, F.R. Niemann-Pick type C disease: Molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011, 116, 789–795. [Google Scholar] [CrossRef]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Lai, M.M. Hepatitis C virus egress and release depend on endosomal trafficking of core protein. J. Virol. 2010, 84, 11590–11598. [Google Scholar] [CrossRef]
- Poh, M.K.; Shui, G.; Xie, X.; Shi, P.Y.; Wenk, M.R.; Gu, F. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antivir. Res. 2012, 93, 191–198. [Google Scholar]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hofmann-Winkler, H.; Kaup, F.; Pöhlmann, S. Host Cell Factors in Filovirus Entry: Novel Players, New Insights. Viruses 2012, 4, 3336-3362. https://doi.org/10.3390/v4123336
Hofmann-Winkler H, Kaup F, Pöhlmann S. Host Cell Factors in Filovirus Entry: Novel Players, New Insights. Viruses. 2012; 4(12):3336-3362. https://doi.org/10.3390/v4123336
Chicago/Turabian StyleHofmann-Winkler, Heike, Franziska Kaup, and Stefan Pöhlmann. 2012. "Host Cell Factors in Filovirus Entry: Novel Players, New Insights" Viruses 4, no. 12: 3336-3362. https://doi.org/10.3390/v4123336
APA StyleHofmann-Winkler, H., Kaup, F., & Pöhlmann, S. (2012). Host Cell Factors in Filovirus Entry: Novel Players, New Insights. Viruses, 4(12), 3336-3362. https://doi.org/10.3390/v4123336