Innate Immunity Evasion by Dengue Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
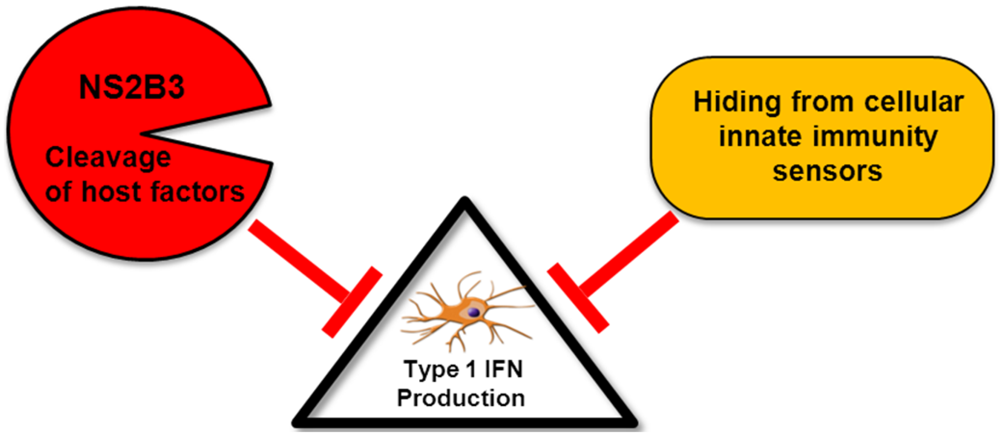
2.1. Inhibition of Type I IFN Production by DENV

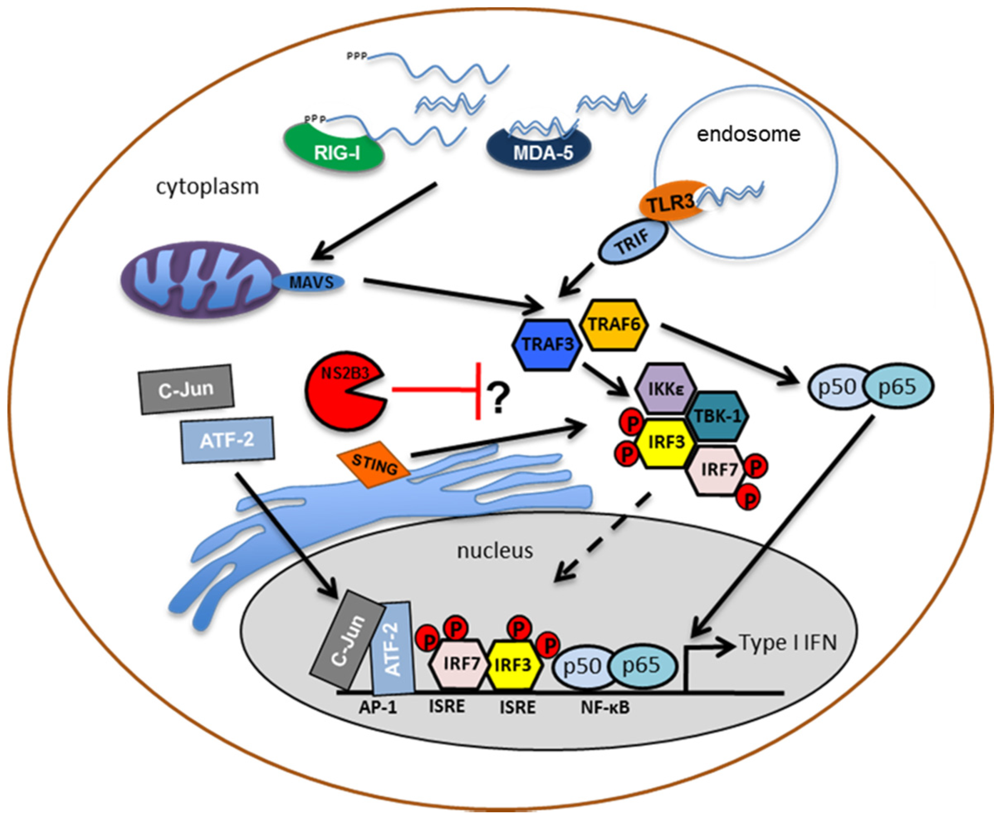
2.1.1. Passive Evasion of PRR Detection
2.1.2. Active Inhibition of Type I IFN Production by DENV

2.2. Inhibition of Type I IFN Signaling by DENV

2.2.1. Pretreatment with Type I IFN Can Inhibit DENV
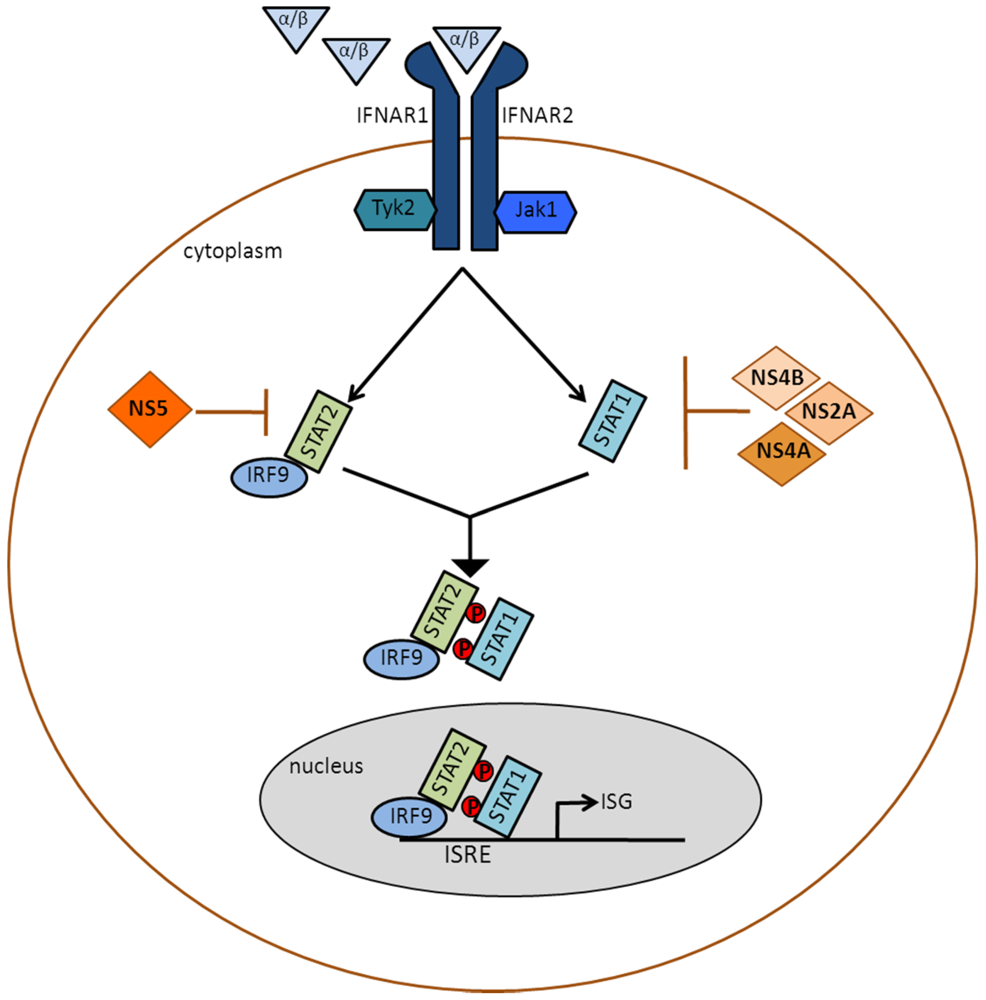
2.2.2. DENV Inhibits Type I IFN Signaling
2.2.2.1. NS2A-, NS4A- and NS4B-Mediated Inhibition of STAT1 Phosphorylation
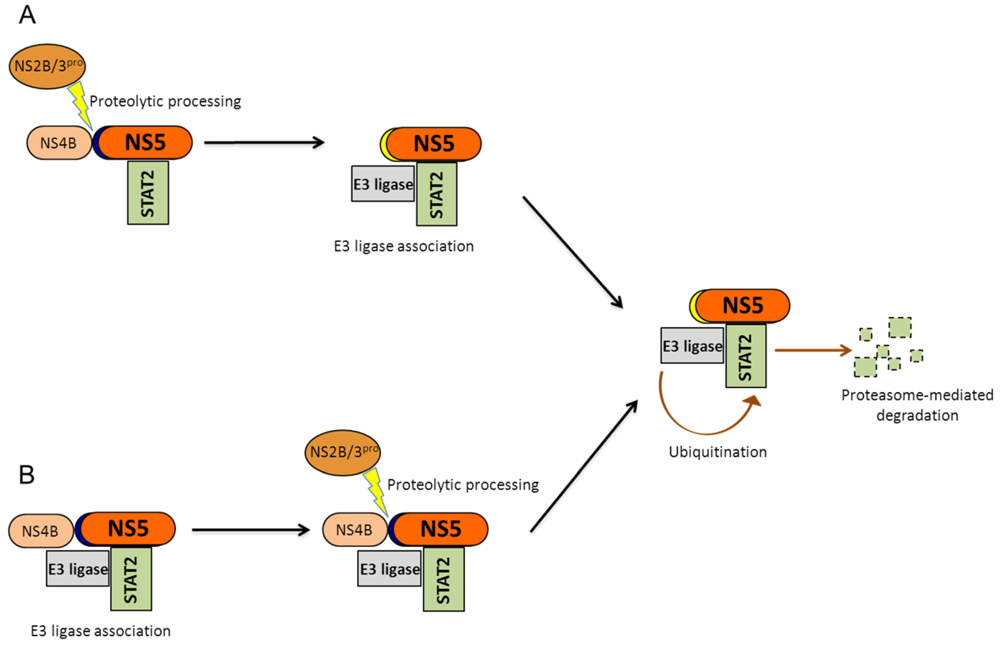
2.2.2.2. NS5-Mediated Degradation of STAT2

2.2.3. Type I IFN Contributes to the Limited Host Tropism of DENV
3. General Conclusions
Acknowledgments
Conflict of Interest
References
- Ackermann, M.; Padmanabhan, R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 2001, 276, 39926–39937. [Google Scholar]
- Alvarez, D.E.; de Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Paludan, S.R. IFN-lambda: Novel antiviral cytokines. J. Interferon Cytokine Res. 2006, 26, 373–379. [Google Scholar]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; Garcia-Sastre, A. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe 2010, 8, 410–421. [Google Scholar]
- Basler, C.F.; Wang, X.; Muhlberger, E.; Volchkov, V.; Paragas, J.; Klenk, H.D.; Garcia-Sastre, A.; Palese, P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA 2000, 97, 12289–12294. [Google Scholar]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; Adams, D.J.; Xavier, R.J.; Farzan, M.; Elledge, S.J. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; Thiel, V.; Sen, G.C.; Fensterl, V.; Klimstra, W.B.; Pierson, T.C.; Buller, R.M.; Gale, M., Jr.; Shi, P.Y.; Diamond, M.S. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar]
- de Veer, M.J.; Holko, M.; Frevel, M.; Walker, E.; Der, S.; Paranjape, J.M.; Silverman, R.H.; Williams, B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001, 69, 912–920. [Google Scholar]
- den Boon, J.A.; Ahlquist, P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar]
- Diamond, M.S.; Harris, E. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology 2001, 289, 297–311. [Google Scholar]
- Diamond, M.S.; Roberts, T.G.; Edgil, D.; Lu, B.; Ernst, J.; Harris, E. Modulation of Dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 2000, 74, 4957–4966. [Google Scholar]
- Dong, H.; Chang, D.C.; Xie, X.; Toh, Y.X.; Chung, K.Y.; Zou, G.; Lescar, J.; Lim, S.P.; Shi, P.Y. Biochemical and genetic characterization of dengue virus methyltransferase. Virology 2010, 405, 568–578. [Google Scholar]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar]
- Evans, J.D.; Seeger, C. Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar]
- Fernandez-Sesma, A.; Marukian, S.; Ebersole, B.J.; Kaminski, D.; Park, M.S.; Yuen, T.; Sealfon, S.C.; Garcia-Sastre, A.; Moran, T.M. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 2006, 80, 6295–6304. [Google Scholar]
- Fredericksen, B.L.; Gale, M., Jr. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; Sulkowski, M.; McHutchison, J.G.; Goldstein, D.B. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar]
- Hidmark, A.S.; McInerney, G.M.; Nordstrom, E.K.; Douagi, I.; Werner, K.M.; Liljestrom, P.; Karlsson Hedestam, G.B. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J. Virol. 2005, 79, 10376–10385. [Google Scholar]
- Ho, L.J.; Hung, L.F.; Weng, C.Y.; Wu, W.L.; Chou, P.; Lin, Y.L.; Chang, D.M.; Tai, T.Y.; Lai, J.H. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J. Immunol. 2005, 174, 8163–8172. [Google Scholar]
- Ho, L.J.; Wang, J.J.; Shaio, M.F.; Kao, C.L.; Chang, D.M.; Han, S.W.; Lai, J.H. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J. Immunol. 2001, 166, 1499–1506. [Google Scholar]
- Iglesias, N.G.; Filomatori, C.V.; Gamarnik, A.V. F1 motif of dengue virus polymerase NS5 is involved in promoter-dependent RNA synthesis. J. Virol. 2011. [Google Scholar]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar]
- Jessie, K.; Fong, M.Y.; Devi, S.; Lam, S.K.; Wong, K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004, 189, 1411–1418. [Google Scholar]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.Y.; Block, T.M.; Guo, J.T. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar]
- Johnson, A.J.; Roehrig, J.T. New mouse model for dengue virus vaccine testing. J. Virol. 1999, 73, 783–786. [Google Scholar]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar]
- King, A.D.; Nisalak, A.; Kalayanrooj, S.; Myint, K.S.; Pattanapanyasat, K.; Nimmannitya, S.; Innis, B.L. B cells are the principal circulating mononuclear cells infected by dengue virus. Southeast Asian J. Trop. Med. Public Health 1999, 30, 718–728. [Google Scholar]
- Kou, Z.; Quinn, M.; Chen, H.; Rodrigo, W.W.; Rose, R.C.; Schlesinger, J.J.; Jin, X. Monocytes, but not T or B cells, are the principal target cells for dengue virus (DV) infection among human peripheral blood mononuclear cells. J. Med. Virol. 2008, 80, 134–146. [Google Scholar] [CrossRef]
- Kraus, T.A.; Garza, L.; Horvath, C.M. Enabled interferon signaling evasion in an immune-competent transgenic mouse model of parainfluenza virus 5 infection. Virology 2008, 371, 196–205. [Google Scholar]
- Kummerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; Barrett, A.D.; Mason, P.W.; Bloom, M.E.; Garcia-Sastre, A.; Khromykh, A.A.; Best, S.M. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar]
- Leung, J.Y.; Pijlman, G.P.; Kondratieva, N.; Hyde, J.; Mackenzie, J.M.; Khromykh, A.A. Role of nonstructural protein NS2A in flavivirus assembly. J. Virol. 2008, 82, 4731–4741. [Google Scholar]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R.; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; Gale, M., Jr. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar]
- Nasirudeen, A.M.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.P.; Liu, D.X. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar]
- Nomaguchi, M.; Ackermann, M.; Yon, C.; You, S.; Padmanabhan, R. De novo synthesis of negative-strand RNA by Dengue virus RNA-dependent RNA polymerase in vitro: Nucleotide, primer, and template parameters. J. Virol. 2003, 77, 8831–8842. [Google Scholar]
- Parisien, J.P.; Lau, J.F.; Horvath, C.M. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J. Virol. 2002, 76, 6435–6441. [Google Scholar]
- Park, C.; Lecomte, M.J.; Schindler, C. Murine Stat2 is uncharacteristically divergent. Nucleic Acids Res. 1999, 27, 4191–4199. [Google Scholar]
- Park, G.S.; Morris, K.L.; Hallett, R.G.; Bloom, M.E.; Best, S.M. Identification of residues critical for the interferon antagonist function of Langat virus NS5 reveals a role for the RNA-dependent RNA polymerase domain. J. Virol. 2007, 81, 6936–6946. [Google Scholar]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7. [Google Scholar]
- Rico-Hesse, R. Dengue virus virulence and transmission determinants. Curr. Top. Microbiol. Immunol. 2011, 338, 45–55. [Google Scholar]
- Rodriguez-Madoz, J.R.; Belicha-Villanueva, A.; Bernal-Rubio, D.; Ashour, J.; Ayllon, J.; Fernandez-Sesma, A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 2010, 84, 9760–9774. [Google Scholar]
- Rodriguez-Madoz, J.R.; Bernal-Rubio, D.; Kaminski, D.; Boyd, K.; Fernandez-Sesma, A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 2010, 84, 4845–4850. [Google Scholar]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar]
- Sun, P.; Fernandez, S.; Marovich, M.A.; Palmer, D.R.; Celluzzi, C.M.; Boonnak, K.; Liang, Z.; Subramanian, H.; Porter, K.R.; Sun, W.; Burgess, T.H. Functional characterization of ex vivo blood myeloid and plasmacytoid dendritic cells after infection with dengue virus. Virology 2009, 383, 207–215. [Google Scholar]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; Riordan, S.; Sheridan, D.; Smedile, A.; Fragomeli, V.; Muller, T.; Bahlo, M.; Stewart, G.J.; Booth, D.R.; George, J. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar]
- Tan, B.H.; Fu, J.; Sugrue, R.J.; Yap, E.H.; Chan, Y.C.; Tan, Y.H. Recombinant dengue type 1 virus NS5 protein expressed in Escherichia coli exhibits RNA-dependent RNA polymerase activity. Virology 1996, 216, 317–325. [Google Scholar]
- Thomas, D.L.; Thio, C.L.; Martin, M.P.; Qi, Y.; Ge, D.; O’Huigin, C.; Kidd, J.; Kidd, K.; Khakoo, S.I.; Alexander, G.; Goedert, J.J.; Kirk, G.D.; Donfield, S.M.; Rosen, H.R.; Tobler, L.H.; Busch, M.P.; McHutchison, J.G.; Goldstein, D.B.; Carrington, M. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009, 461, 798–801. [Google Scholar]
- Tsai, Y.T.; Chang, S.Y.; Lee, C.N.; Kao, C.L. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Umareddy, I.; Tang, K.F.; Vasudevan, S.G.; Devi, S.; Hibberd, M.L.; Gu, F. Dengue virus regulates type I interferon signalling in a strain-dependent manner in human cell lines. J. Gen. Virol. 2008, 89, 3052–3062. [Google Scholar]
- Villordo, S.M.; Alvarez, D.E.; Gamarnik, A.V. A balance between circular and linear forms of the dengue virus genome is crucial for viral replication. RNA 2010, 16, 2325–2335. [Google Scholar]
- Warke, R.V.; Martin, K.J.; Giaya, K.; Shaw, S.K.; Rothman, A.L.; Bosch, I. TRAIL is a novel antiviral protein against dengue virus. J. Virol. 2008, 82, 555–564. [Google Scholar]
- Welsch, C.; Zeuzem, S. RNA-binding activity of hepatitis C virus NS4B: A novel target for small molecule inhibitors. Gastroenterology 2009, 137, 2170–2172. [Google Scholar]
- Werme, K.; Wigerius, M.; Johansson, M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 2008, 10, 696–712. [Google Scholar]
- Wu, S.J.; Grouard-Vogel, G.; Sun, W.; Mascola, J.R.; Brachtel, E.; Putvatana, R.; Louder, M.K.; Filgueira, L.; Marovich, M.A.; Wong, H.K.; Blauvelt, A.; Murphy, G.S.; Robb, M.L.; Innes, B.L.; Birx, D.L.; Hayes, C.G.; Frankel, S.S. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000, 6, 816–820. [Google Scholar]
- Yauch, L.E.; Shresta, S. Mouse models of dengue virus infection and disease. Antiviral. Res. 2008, 80, 87–93. [Google Scholar]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar]
- Zou, G.; Chen, Y.L.; Dong, H.; Lim, C.C.; Yap, L.J.; Yau, Y.H.; Shochat, S.G.; Lescar, J.; Shi, P.Y. Functional Analysis of Two Cavities in Flavivirus NS5 Polymerase. J. Biol. Chem. 2011, 286, 14362–14372. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morrison, J.; Aguirre, S.; Fernandez-Sesma, A. Innate Immunity Evasion by Dengue Virus. Viruses 2012, 4, 397-413. https://doi.org/10.3390/v4030397
Morrison J, Aguirre S, Fernandez-Sesma A. Innate Immunity Evasion by Dengue Virus. Viruses. 2012; 4(3):397-413. https://doi.org/10.3390/v4030397
Chicago/Turabian StyleMorrison, Juliet, Sebastian Aguirre, and Ana Fernandez-Sesma. 2012. "Innate Immunity Evasion by Dengue Virus" Viruses 4, no. 3: 397-413. https://doi.org/10.3390/v4030397
APA StyleMorrison, J., Aguirre, S., & Fernandez-Sesma, A. (2012). Innate Immunity Evasion by Dengue Virus. Viruses, 4(3), 397-413. https://doi.org/10.3390/v4030397