2.1. The Ectodomains of Gp2b, Gp3 and Gp4 Are Insoluble When Expressed in E. coli
To express the EAV-glycoproteins in
E. coli in a form likely to be soluble, it might be necessary to delete hydrophobic domains. Hence, we performed Kyte-Doolittle plots with gp2b, gp3 and gp4 to identify their hydrophobic regions [
17]. Surprisingly, besides the described N- and C-terminal hydrophobic domains, which are likely to function as signal peptide or transmembrane region, all three glycoproteins contain another hydrophobic region in the middle of the molecule (
Figure 1A). This pattern of hydrophobic domains is reminiscent of viral fusion proteins, such as the hemagglutinin of influenza virus, where the third hydrophobic domain functions as a fusion peptide, which is exposed at the surface of the molecule and inserts into the cellular membrane catalysing its fusion with the viral membrane [
18,
19]. The occurrence of three hydrophobic domains in each EAV-glycoprotein suggests that each contains a fusion peptide and thus supports the concept that the gp2b/gp3/gp4-complex is involved in membrane fusion.
We cloned DNA sequences encoding the ectodomains of gp2b, gp3 and gp4 into pQE expression-plasmids. The resulting constructs contained a 6xHis affinity tag instead of the N-terminal signal peptide and lacked the C-terminal transmembrane region. Expression of His-gp2b and His-gp4 in
E. coli was clearly detectable by SDS-PAGE and Coomassie-staining of cellular extracts (
Figure 1B), but His-gp3 was not expressed at all (data not shown). When cellular extracts were separated by centrifugation into a soluble and a particulate fraction prior to SDS-PAGE, it became obvious that both His-gp2b and His-gp4 are completely insoluble. We therefore replaced the N-terminal histidines by glutathione-S-transferase (Gst), which is supposed to increase the solubility of proteins to which it is attached. Nevertheless, Gst-gp2b and Gst-gp4 were still exclusively present in the particulate fraction of
E. coli. However, fusion to Gst instead to 6xHis now allows expression of gp3, but Gst-gp3 was also completely insoluble (
Figure 1B). Several attempts to enhance the solubility of the EAV-proteins,
i.e., by reducing the expression time or temperature or the amount of IPTG used for induction, were not successful (not shown). Thus, all three glycoproteins do not fold into a soluble conformation when expressed in
E. coli, but instead are deposited as insoluble aggregates in inclusion bodies. However, recent data suggested that inclusion bodies do not necessarily contain “useless”,
i.e., misfolded proteins, but proteins present in alternative conformational states [
20,
21].
2.2. Refolding of the Ectodomains of Gp2b, Gp3 and Gp4 from Inclusion Bodies
The inclusion bodies were extracted with denaturating agents (2 M urea, pH 12.5) to dissolve the aggregates and unfold the proteins. To (possibly) allow refolding of proteins [
22], incompletely solubilized components were pelleted and the resulting supernatant was subjected to stepwise dialysis against buffers containing decreasing urea concentration and finally against physiological phosphate-buffer. To analyze whether the solubilized protein were precipitated or remained soluble during this procedure, each dialysate was centrifuged and the resulting supernatant and pellet were subjected to SDS-PAGE. Whereas His-gp2b and His-gp4 completely precipitated during dialysis, Gst-gp3 and Gst‑gp4 remained soluble indicating that the Gst-tag facilitated folding of the proteins into a soluble conformation (
Figure 2, upper panel). In contrast, refolding of GST-Gp2b was very inefficient (not shown).
Figure 1.
Expression of the ectodomains of gp2b, gp3 and gp4 in E. coli and their accumulation in inclusion bodies. (A) Kyte-Doolittle hydropathy plots (window size 9) for gp2b, gp3, gp4 and for the hemagglutinin of influenza virus (Flu).The hydrophobicity score is plotted against the amino acid residue (position) of the respective protein. Scores above 2 are considered to be highly hydrophobic, i.e., are likely to insert into membranes. The N- and C-terminal hydrophobic regions are the proposed signal peptides and the transmembrane regions, respectively. The third hydrophobic region in the middle of the molecule, which has been shown to function as fusion peptide in the case of HA, is marked with a white arrow. The constructs used in this study contain the complete ectodomain of gp2b, gp3 and gp4, i.e., excluding the N- and C-terminal hydrophobic region. (B) Expression of 6 × His-tagged (upper panel) and Gst-EAV-proteins (lower panel) in E.coli and their accumulation in inclusion bodies. E. coli containing the respective expression plasmids were induced with IPTG and were grown overnight at room temperature. Pelleted cells were resuspended in lysis-buffer, lysed by sonication and centrifuged for 20 min at 5,000 × g. Aliquots of cell lysates before (–) and after (+) induction of protein synthesis as well as supernatants (S) and pellets (P) were subjected to SDS‑PAGE under reducing conditions and Coomassie staining. The expressed EAV‑proteins are marked with a black arrow. M: molecular mass markers as indicated, Ly: Lysozyme (14 kDa), which is added before preparation of cell lysates.
Figure 1.
Expression of the ectodomains of gp2b, gp3 and gp4 in E. coli and their accumulation in inclusion bodies. (A) Kyte-Doolittle hydropathy plots (window size 9) for gp2b, gp3, gp4 and for the hemagglutinin of influenza virus (Flu).The hydrophobicity score is plotted against the amino acid residue (position) of the respective protein. Scores above 2 are considered to be highly hydrophobic, i.e., are likely to insert into membranes. The N- and C-terminal hydrophobic regions are the proposed signal peptides and the transmembrane regions, respectively. The third hydrophobic region in the middle of the molecule, which has been shown to function as fusion peptide in the case of HA, is marked with a white arrow. The constructs used in this study contain the complete ectodomain of gp2b, gp3 and gp4, i.e., excluding the N- and C-terminal hydrophobic region. (B) Expression of 6 × His-tagged (upper panel) and Gst-EAV-proteins (lower panel) in E.coli and their accumulation in inclusion bodies. E. coli containing the respective expression plasmids were induced with IPTG and were grown overnight at room temperature. Pelleted cells were resuspended in lysis-buffer, lysed by sonication and centrifuged for 20 min at 5,000 × g. Aliquots of cell lysates before (–) and after (+) induction of protein synthesis as well as supernatants (S) and pellets (P) were subjected to SDS‑PAGE under reducing conditions and Coomassie staining. The expressed EAV‑proteins are marked with a black arrow. M: molecular mass markers as indicated, Ly: Lysozyme (14 kDa), which is added before preparation of cell lysates.
![Viruses 04 00414 g001]()
Transmembrane proteins forming a heterooligomeric spike in virus particles often require each other for proper folding inside cells [
14,
15]. To analyze whether folding of His-gp2b and/or His-gp4 might be positively affected by the presence of Gst-gp3 or Gst-gp4
in vitro, we combined various supernatants obtained from the urea extracts of inclusion bodies prior to dialysis. Strikingly, His-gp2b as well as His-gp4 remain (for most parts or even completely) soluble when dialyzed together with Gst-gp3 or with Gst-gp4 (
Figure 2, lower panel). Thus, His-gp4 as well as His-gp2b can fold into a soluble conformation when either Gst-gp3 or Gst-gp4 is present during dialysis.
Figure 2.
Refolding of EAV proteins during dialysis of urea-extracts of inclusion bodies. Upper panel: Dialysis of individual EAV-proteins. Lower panel: Combined dialysis of the indicated combinations of EAV-proteins. Inclusion bodies were resuspended in denaturating buffer. After centrifugation the resulting supernatants were dialyzed against buffer with decreasing urea concentration and finally against physiological buffer. The dialysate was then centrifuged and aliquots of the supernatant (S) and of the pellet (P) were analyzed by SDS-PAGE under reducing conditions and Coomassie-staining. M: molecular mass marker as indicated, Ly: Lysozyme. Due to the lack of antibodies we were not able to confirm the identiy of the bands by Western blot. Nevertheless, since the recombinant proteins are the major proteins present in bacterial lysates (see
Figure 1), we are sure that the identification of bands is correct.
Figure 2.
Refolding of EAV proteins during dialysis of urea-extracts of inclusion bodies. Upper panel: Dialysis of individual EAV-proteins. Lower panel: Combined dialysis of the indicated combinations of EAV-proteins. Inclusion bodies were resuspended in denaturating buffer. After centrifugation the resulting supernatants were dialyzed against buffer with decreasing urea concentration and finally against physiological buffer. The dialysate was then centrifuged and aliquots of the supernatant (S) and of the pellet (P) were analyzed by SDS-PAGE under reducing conditions and Coomassie-staining. M: molecular mass marker as indicated, Ly: Lysozyme. Due to the lack of antibodies we were not able to confirm the identiy of the bands by Western blot. Nevertheless, since the recombinant proteins are the major proteins present in bacterial lysates (see
Figure 1), we are sure that the identification of bands is correct.
2.3. Oligomerization Is Required for Refolding of the Ectodomains of Gp2b, Gp3 and Gp4
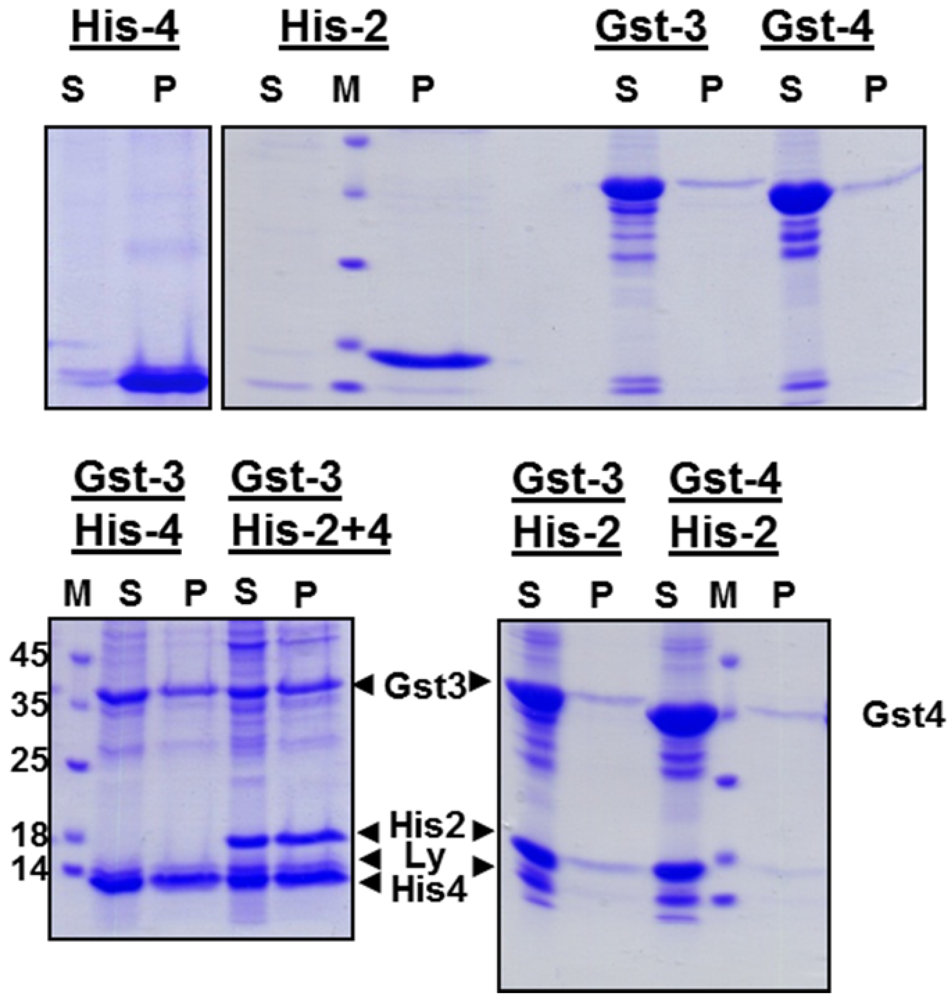
We tested whether oligomerization of EAV-proteins occurs during refolding. After dialysis of Gst‑gp3, either alone or in combination with His-gp2b, or with His-gp4, or with both proteins, the cleared dialysate was loaded onto a glutathione-column, the column was washed, and bound proteins were eluted with glutathione. SDS-PAGE of these fractions shows that the His-tagged proteins are present together with Gst-gp3 in the eluate for all combinations (
Figure 3). This must be due to the formation of oligomers during refolding, since lysozyme, which is added before lysis of bacteria, is present in the fraction which was loaded onto the column, but largely removed during affinity chromatography. However, it cannot be inferred from the data of the experiments involving all three EAV-proteins, whether a heterotrimeric complex or two heterodimers,
i.e., Gst-gp3/His-gp2b and Gst‑gp3/His-gp4, have been formed. Since the complete trimeric Gp2b/3/4 complex is very unstable, e.g., it does not resist purification by centrifugation through a sucrose gradient [
12], we were not able to clarify this important question.
Figure 3.
Affinity-purification of EAV-protein oligomers. Gst-gp3, His-gp2b and His-gp4 were expressed individually in E. coli and solubilized from inclusion bodies, and refolded by dialysis. Urea extracts from inclusion bodies containing Gst-gp3 were dialysed alone (Gst-3) or were combined with His-gp2b (Gst-3+His-2b), with His-gp4 (Gst-3His-4) or with both (Gst-3+His-2b+4) prior to dialysis. The cleared dialysate was then agitated with glutathione-beads, beads were pelleted, put in a column, and washed 5 times with phosphate-buffered saline. Proteins were eluted with elution buffer. Aliquots of the dialysate (load), of the flow-through (FT), of wash 5 (W5) and of the eluates (E) were subjected to SDS-PAGE under reducing conditions and Coomassie-staining. M: Molecular mass markers, Ly: Lysozyme.
Figure 3.
Affinity-purification of EAV-protein oligomers. Gst-gp3, His-gp2b and His-gp4 were expressed individually in E. coli and solubilized from inclusion bodies, and refolded by dialysis. Urea extracts from inclusion bodies containing Gst-gp3 were dialysed alone (Gst-3) or were combined with His-gp2b (Gst-3+His-2b), with His-gp4 (Gst-3His-4) or with both (Gst-3+His-2b+4) prior to dialysis. The cleared dialysate was then agitated with glutathione-beads, beads were pelleted, put in a column, and washed 5 times with phosphate-buffered saline. Proteins were eluted with elution buffer. Aliquots of the dialysate (load), of the flow-through (FT), of wash 5 (W5) and of the eluates (E) were subjected to SDS-PAGE under reducing conditions and Coomassie-staining. M: Molecular mass markers, Ly: Lysozyme.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


