Evolution of Foamy Viruses: The Most Ancient of All Retroviruses †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
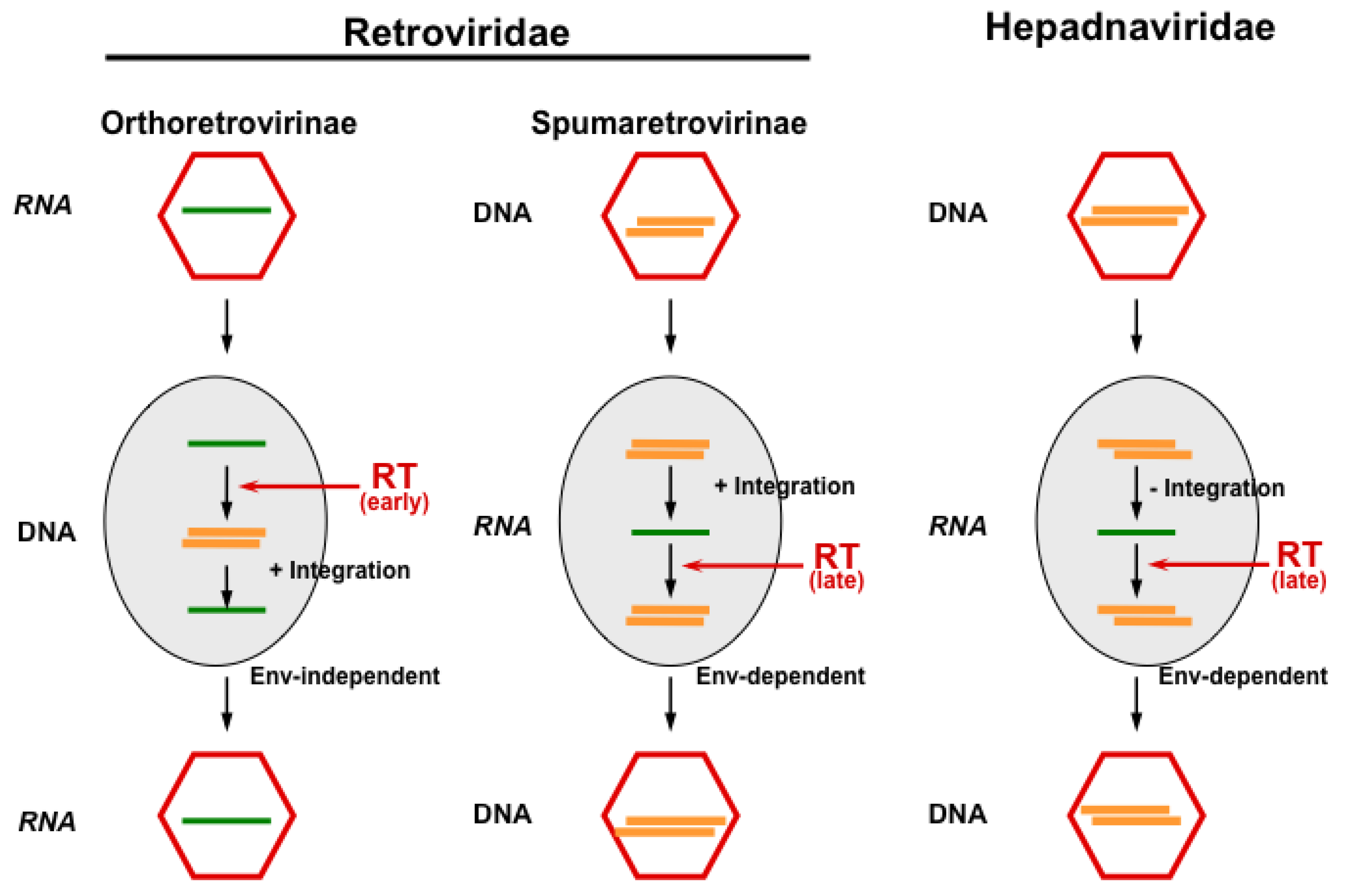
:1. Introduction
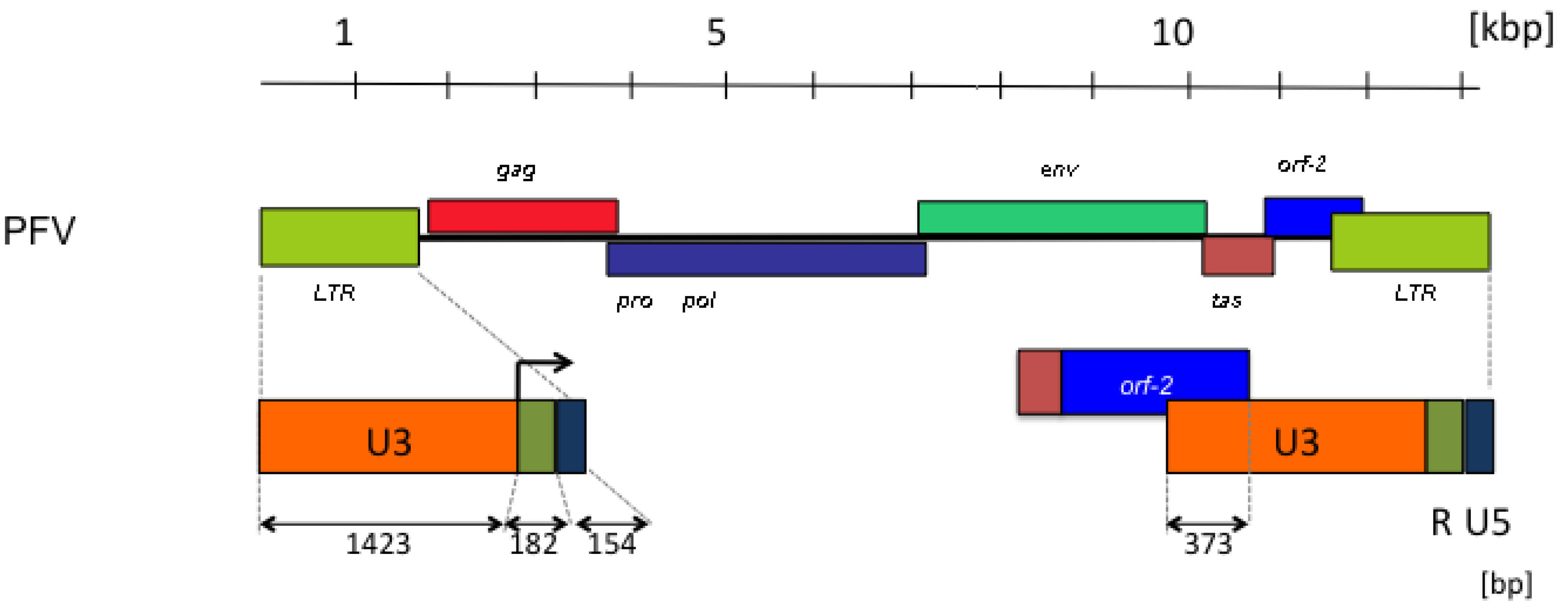
2. FV Sequence Conservation


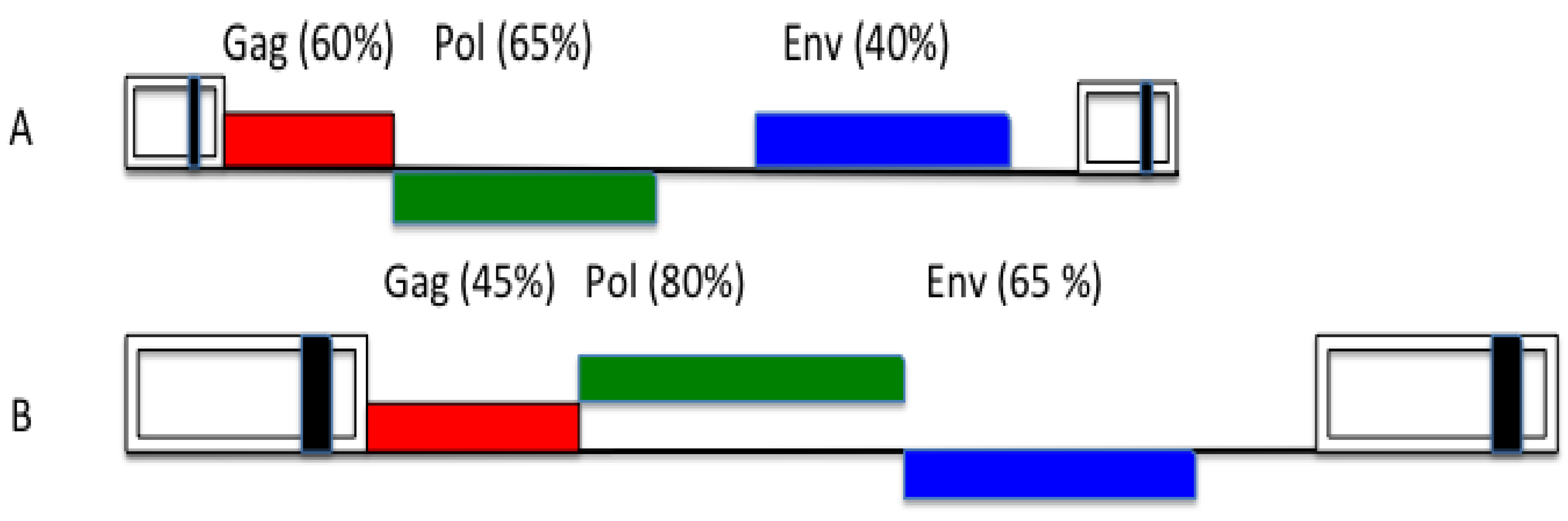
3. FV Recombination
4. Retroviral Genome Diversity

5. Innate Host Defense and Viral Counter-Defense
6. On the Origin of PFV and Further Human FV Infections


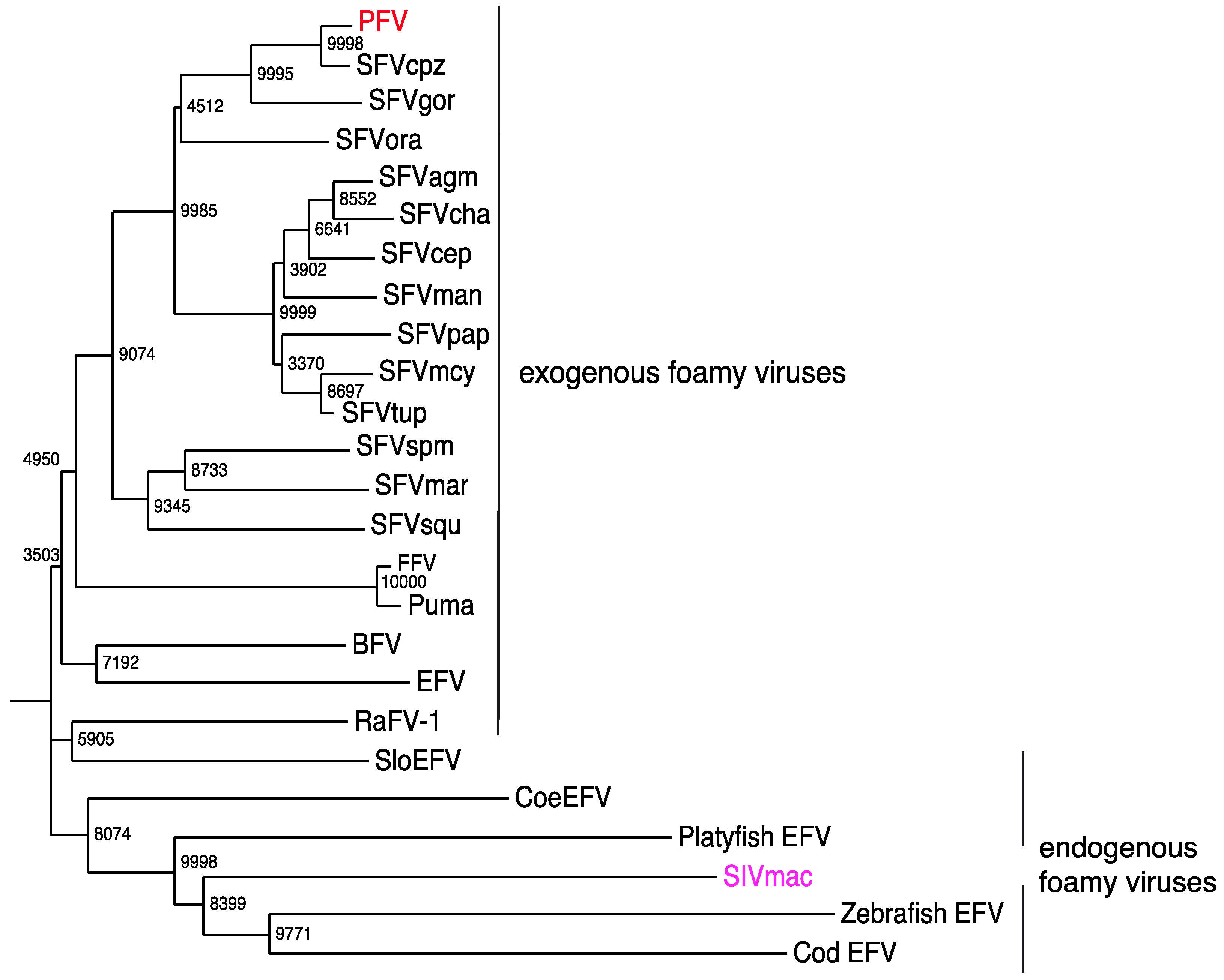
7. Exogenous FVs in the Animal Kingdom
8. Endogenous FVs
9. Conclusions
Acknowledgements
Conflicts of Interest
References
- Linial, M.L. Foamy viruses are unconventional retroviruses. J. Virol. 1999, 73, 1747–1755. [Google Scholar]
- Rethwilm, A. Molecular biology of foamy viruses. Med. Microbiol. Immunol. 2010, 199, 197–207. [Google Scholar] [CrossRef]
- Rethwilm, A.; Lindemann, D. Foamy Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 6, pp. 1613–1632. [Google Scholar]
- Lindemann, D.; Rethwilm, A. Foamy virus biology and its application for vector development. Viruses 2011, 3, 561–585. [Google Scholar] [CrossRef]
- Rethwilm, A. Foamy virus vectors: An awaited alternative to gammaretro- and lentiviral vectors. Curr. Gene Ther. 2007, 7, 261–271. [Google Scholar] [CrossRef]
- Trobridge, G.D. Foamy virus vectors for gene transfer. Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Maertens, G.N.; Hare, S.; Cherepanov, P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 2010, 468, 326–329. [Google Scholar] [CrossRef]
- Valkov, E.; Gupta, S.S.; Hare, S.; Helander, A.; Roversi, P.; McClure, M.; Cherepanov, P. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res. 2009, 37, 243–255. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J.J. The Origin and Evolution of Viruses. In Virology; Mahe, B.W.J., ter Meulen, V., Eds.; Edward Arnold: London, UK, 2005; Volume 1, pp. 11–23. [Google Scholar]
- Domingo, E. Virus Evolution. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams: Philadelphia, PA, USA, 2007; Volume 5, pp. 389–421. [Google Scholar]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev.: MMBR 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: Pathogenetic and therapeutic implications. J. Virol. 2007, 81, 10209–10219. [Google Scholar] [CrossRef]
- Korber, B.T.; Letvin, N.L.; Haynes, B.F. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J. Virol. 2009, 83, 8300–8314. [Google Scholar] [CrossRef]
- Osiowy, C.; Giles, E.; Tanaka, Y.; Mizokami, M.; Minuk, G.Y. Molecular evolution of hepatitis B virus over 25 years. J. Virol. 2006, 80, 10307–10314. [Google Scholar] [CrossRef]
- Simmonds, P.; Mellor, J.; Sakuldamrongpanich, T.; Nuchaprayoon, C.; Tanprasert, S.; Holmes, E.C.; Smith, D.B. Evolutionary analysis of variants of hepatitis C virus found in South-East Asia: Comparison with classifications based upon sequence similarity. J. Gen. Virol. 1996, 77, 3013–3024. [Google Scholar] [CrossRef]
- Andernach, I.E.; Jutavijittum, P.; Samountry, B.; Yousukh, A.; Thammavong, T.; Hubschen, J.M.; Muller, C.P. A high variability of mixed infections and recent recombinations of hepatitis B virus in Laos. PLoS One 2012, 7, e30245. [Google Scholar] [CrossRef]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 2011, 25, 679–689. [Google Scholar] [CrossRef]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus--15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef]
- Wang, G.P.; Sherrill-Mix, S.A.; Chang, K.M.; Quince, C.; Bushman, F.D. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J. Virol. 2010, 84, 6218–6228. [Google Scholar] [CrossRef]
- Zhang, M.; Foley, B.; Schultz, A.K.; Macke, J.P.; Bulla, I.; Stanke, M.; Morgenstern, B.; Korber, B.; Leitner, T. The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology 2010, 7, 25. [Google Scholar] [CrossRef]
- Shi, W.; Carr, M.J.; Dunford, L.; Zhu, C.; Hall, W.W.; Higgins, D.G. Identification of novel inter-genotypic recombinants of human hepatitis B viruses by large-scale phylogenetic analysis. Virology 2012, 427, 51–59. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef]
- Schweizer, M.; Neumann-Haefelin, D. Phylogenetic analysis of primate foamy viruses by comparison of pol sequences. Virology 1995, 207, 577–582. [Google Scholar] [CrossRef]
- Schweizer, M.; Schleer, H.; Pietrek, M.; Liegibel, J.; Falcone, V.; Neumann-Haefelin, D. Genetic stability of foamy viruses: Long-term study in an African green monkey population. J. Virol. 1999, 73, 9256–9265. [Google Scholar]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef]
- Verschoor, E.J.; Langenhuijzen, S.; Bontjer, I.; Fagrouch, Z.; Niphuis, H.; Warren, K.S.; Eulenberger, K.; Heeney, J.L. The phylogeography of orangutan foamy viruses supports the theory of ancient repopulation of Sumatra. J. Virol. 2004, 78, 12712–12716. [Google Scholar] [CrossRef]
- Blasse, A.; Calvignac-Spencer, S.; Merkel, K.; Goffe, A.S.; Boesch, C.; Mundry, R.; Leendertz, F.H. Mother-offspring transmission and age-dependent accumulation of simian foamy virus in wild chimpanzees. J. Virol. 2013, 87, 5193–5204. [Google Scholar] [CrossRef]
- Mason, W.S.; Burrell, C.J.; Casey, J.; Gerlich, W.H.; Howard, C.R.; Kann, M.; Lanford, R.; Newbold, J.; Schaefer, S.; Taylor, J.M.; et al. Hepadnaviruses. In Virus Taxonomy; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: London, UK, 2005; pp. 373–384. [Google Scholar]
- Miller, R.H.; Robinson, W.S. Common evolutionary origin of hepatitis B virus and retroviruses. Proc. Natl. Acad. Sci. USA 1986, 83, 2531–2535. [Google Scholar] [CrossRef]
- Zlotnick, A.; Stahl, S.J.; Wingfield, P.T.; Conway, J.F.; Cheng, N.; Steven, A.C. Shared motifs of the capsid proteins of hepadnaviruses and retroviruses suggest a common evolutionary origin. FEBS Lett. 1998, 431, 301–304. [Google Scholar] [CrossRef]
- Enssle, J.; Jordan, I.; Mauer, B.; Rethwilm, A. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc. Natl. Acad. Sci. USA 1996, 93, 4137–4141. [Google Scholar] [CrossRef]
- Lecellier, C.H.; Saib, A. Foamy viruses: Between retroviruses and pararetroviruses. Virology 2000, 271, 1–8. [Google Scholar] [CrossRef]
- Rethwilm, A. Unexpected replication pathways of foamy viruses. J. Acquir Immune Defic. Syndr. Hum. Retrovirol. 1996, 13 (Suppl. 1), S248–S253. [Google Scholar] [CrossRef]
- Liu, W.; Worobey, M.; Li, Y.; Keele, B.F.; Bibollet-Ruche, F.; Guo, Y.; Goepfert, P.A.; Santiago, M.L.; Ndjango, J.B.; Neel, C.; et al. Molecular ecology and natural history of simian foamy virus infection in wild-living chimpanzees. PLoS Pathog. 2008, 4, e1000097. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- Rinke, C.S.; Boyer, P.L.; Sullivan, M.D.; Hughes, S.H.; Linial, M.L. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J. Virol. 2002, 76, 7560–7570. [Google Scholar] [CrossRef]
- Boyer, P.L.; Stenbak, C.R.; Hoberman, D.; Linial, M.L.; Hughes, S.H. In vitro fidelity of the prototype primate foamy virus (PFV) RT compared to HIV-1 RT. Virology 2007, 367, 253–264. [Google Scholar] [CrossRef]
- Schmidt, M.; Herchenröder, O.; Heeney, J.; Rethwilm, A. Long terminal repeat U3 length polymorphism of human foamy virus. Virology 1997, 230, 167–178. [Google Scholar] [CrossRef]
- De Celis, J.; Tobaly-Tapiero, J.; Hampe, A.; Emanoil-Ravier, R. Structure and function of the long terminal repeat of the chimpanzee foamy virus isolates (SFV-6). Arch. Virol. 1994, 138, 345–355. [Google Scholar] [CrossRef]
- Kang, Y.; Blair, W.S.; Cullen, B.R. Identification and functional characterization of a high-affinity Bel-1 DNA binding site located in the human foamy virus internal promoter. J. Virol. 1998, 72, 504–511. [Google Scholar]
- He, F.; Blair, W.S.; Fukushima, J.; Cullen, B.R. The human foamy virus Bel-1 transcription factor is a sequence-specific DNA binding protein. J. Virol. 1996, 70, 3902–3908. [Google Scholar]
- Hartl, M.J.; Bodem, J.; Jochheim, F.; Rethwilm, A.; Rosch, P.; Wöhrl, B.M. Regulation of foamy virus protease activity by viral RNA - a novel and unique mechanism among retroviruses. J. Virol. 2011, 85, 4462–4469. [Google Scholar] [CrossRef]
- Spannaus, R.; Hartl, M.J.; Wohrl, B.M.; Rethwilm, A.; Bodem, J. The prototype foamy virus protease is active independently of the integrase domain. Retrovirology 2012, 9, 41. [Google Scholar] [CrossRef]
- Rethwilm, A.; Darai, G.; Rösen, A.; Maurer, B.; Flügel, R.M. Molecular cloning of the genome of human spumaretrovirus. Gene 1987, 59, 19–28. [Google Scholar] [CrossRef]
- Rua, R.; Betsem, E.; Calattini, S.; Saib, A.; Gessain, A. Genetic characterization of simian foamy viruses infecting humans. J. Virol. 2012, 86, 13350–13359. [Google Scholar] [CrossRef]
- Löchelt, M.; Zentgraf, H.; Flügel, R.M. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology 1991, 184, 43–54. [Google Scholar] [CrossRef]
- Schulze, A.; Lemey, P.; Schubert, J.; McClure, M.O.; Rethwilm, A.; Bodem, J. Complete nucleotide sequence and evolutionary analysis of a gorilla foamy virus. J. Gen. Virol. 2011, 92, 582–586. [Google Scholar] [CrossRef]
- Saib, A.; Koken, M.H.; van der Spek, P.; Peries, J.; de The, H. Involvement of a spliced and defective human foamy virus in the establishment of chronic infection. J. Virol. 1995, 69, 5261–5268. [Google Scholar]
- Saib, A.; Peries, J.; de The, H. A defective human foamy provirus generated by pregenome splicing. EMBO J. 1993, 12, 4439–4444. [Google Scholar]
- Rethwilm, A.; Erlwein, O.; Baunach, G.; Maurer, B.; ter Meulen, V. The transcriptional transactivator of human foamy virus maps to the bel 1 genomic region. Proc. Natl. Acad. Sci. USA 1991, 88, 941–945. [Google Scholar] [CrossRef]
- Gärtner, K.; Wiktorowicz, T.; Park, J.; Mergia, A.; Rethwilm, A.; Scheller, C. Accuracy estimation of foamy virus genome copying. Retrovirology 2009, 6, 32. [Google Scholar] [CrossRef]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schäfer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef]
- Zemba, M.; Alke, A.; Bodem, J.; Winkler, I.G.; Flower, R.L.; Pfrepper, K.; Delius, H.; Flügel, R.M.; Löchelt, M. Construction of infectious feline foamy virus genomes: Cat antisera do not cross-neutralize feline foamy virus chimera with serotype-specific Env sequences. Virology 2000, 266, 150–156. [Google Scholar] [CrossRef]
- Delebecque, F.; Suspene, R.; Calattini, S.; Casartelli, N.; Saib, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef]
- Bock, M.; Heinkelein, M.; Lindemann, D.; Rethwilm, A. Cells expressing the human foamy virus (HFV) accessory Bet protein are resistant to productive HFV superinfection. Virology 1998, 250, 194–204. [Google Scholar] [CrossRef]
- Baunach, G.; Maurer, B.; Hahn, H.; Kranz, M.; Rethwilm, A. Functional analysis of human foamy virus accessory reading frames. J. Virol. 1993, 67, 5411–5418. [Google Scholar]
- Löchelt, M. Foamy virus transactivation and gene expression. Curr. Top. Microbiol. Immunol. 2003, 277, 27–61. [Google Scholar] [CrossRef]
- Muranyi, W.; Flügel, R.M. Analysis of splicing patterns of human spumaretrovirus by polymerase chain reaction reveals complex RNA structures. J. Virol. 1991, 65, 727–735. [Google Scholar]
- Falcone, V.; Leupold, J.; Clotten, J.; Urbanyi, E.; Herchenröder, O.; Spatz, W.; Volk, B.; Bohm, N.; Toniolo, A.; Neumann-Haefelin, D.; et al. Sites of simian foamy virus persistence in naturally infected African green monkeys: Latent provirus is ubiquitous, whereas viral replication is restricted to the oral mucosa. Virology 1999, 257, 7–14. [Google Scholar] [CrossRef]
- Murray, S.M.; Picker, L.J.; Axthelm, M.K.; Hudkins, K.; Alpers, C.E.; Linial, M.L. Replication in a superficial epithelial cell niche explains the lack of pathogenicity of primate foamy virus infections. J. Virol. 2008, 82, 5981–5985. [Google Scholar] [CrossRef]
- Wattel, E.; Cavrois, M.; Gessain, A.; Wain-Hobson, S. Clonal expansion of infected cells: A way of life for HTLV-I. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13 (Suppl. 1), S92–S99. [Google Scholar] [CrossRef]
- Mortreux, F.; Leclercq, I.; Gabet, A.S.; Leroy, A.; Westhof, E.; Gessain, A.; Wain-Hobson, S.; Wattel, E. Somatic mutation in human T-cell leukemia virus type 1 provirus and flanking cellular sequences during clonal expansion in vivo. J. Natl. Cancer Inst. 2001, 93, 367–377. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol. 2012, 3, 388–410. [Google Scholar]
- Khan, A.S.; Kumar, D. Simian foamy virus infection by whole-blood transfer in rhesus macaques: Potential for transfusion transmission in humans. Transfusion 2006, 46, 1352–1359. [Google Scholar] [CrossRef]
- Neumann-Haefelin, D.; Rethwilm, A.; Bauer, G.; Gudat, F.; zur Hausen, H. Characterization of a foamy virus isolated from Cercopithecus aethiops lymphoblastoid cells. Med. Microbiol. Immunol. (Berl.) 1983, 172, 75–86. [Google Scholar] [CrossRef]
- Nora, T.; Charpentier, C.; Tenaillon, O.; Hoede, C.; Clavel, F.; Hance, A.J. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 2007, 81, 7620–7628. [Google Scholar] [CrossRef]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar]
- Switzer, W.M.; Garcia, A.D.; Yang, C.; Wright, A.; Kalish, M.L.; Folks, T.M.; Heneine, W. Coinfection with HIV-1 and simian foamy virus in West Central Africans. J. Infect. Dis. 2008, 197, 1389–1393. [Google Scholar] [CrossRef]
- Galvin, T.A.; Ahmed, I.A.; Shahabuddin, M.; Bryan, T.; Khan, A.S. Identification of Recombination in the Envelope Gene of Simian Foamy Virus Serotype 2 Isolated from Macaca cyclopis. J. Virol. 2013, 87, 8792–8797. [Google Scholar] [CrossRef]
- Hooks, J.J.; Gibbs, C.J., Jr. The foamy viruses. Bacteriol. Rev. 1975, 39, 169–185. [Google Scholar]
- Leendertz, F.H.; Zirkel, F.; Couacy-Hymann, E.; Ellerbrok, H.; Morozov, V.A.; Pauli, G.; Hedemann, C.; Formenty, P.; Jensen, S.A.; Boesch, C.; et al. Interspecies transmission of simian foamy virus in a natural predator-prey system. J. Virol. 2008, 82, 7741–7744. [Google Scholar] [CrossRef]
- Boneva, R.S.; Switzer, W.M.; Spira, T.J.; Bhullar, V.B.; Shanmugam, V.; Cong, M.E.; Lam, L.; Heneine, W.; Folks, T.M.; Chapman, L.E. Clinical and virological characterization of persistent human infection with simian foamy viruses. AIDS Res. Hum. Retrovir. 2007, 23, 1330–1337. [Google Scholar] [CrossRef]
- Roy, J.; Rudolph, W.; Juretzek, T.; Gartner, K.; Bock, M.; Herchenröder, O.; Lindemann, D.; Heinkelein, M.; Rethwilm, A. Feline foamy virus genome and replication strategy. J. Virol. 2003, 77, 11324–11331. [Google Scholar] [CrossRef]
- Rethwilm, A. Foamy Viruses. In Virology; Mahe, B.W.J., ter Meulen, V., Eds.; Edward Arnold: London, UK, 2005; Volume 2, pp. 1304–1321. [Google Scholar]
- Plochmann, K.; Horn, A.; Gschmack, E.; Armbruster, N.; Krieg, J.; Wiktorowicz, T.; Weber, C.; Stirnnagel, K.; Lindemann, D.; Rethwilm, A.; et al. Heparan sulfate is an attachment factor for foamy virus entry. J. Virol. 2012, 86, 10028–10035. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Persons, D.A. Cell Membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol. Ther. 2012, 20, 1158–1166. [Google Scholar] [CrossRef]
- Hunter, E. Viral Entry and Receptors. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, NY 11724, USA, 1997. [Google Scholar]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus Env leader protein with the N-terminal Gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar] [CrossRef]
- Matthes, D.; Wiktorowicz, T.; Zahn, J.; Bodem, J.; Stanke, N.; Lindemann, D.; Rethwilm, A. Basic residues in the foamy virus gag protein. J. Virol. 2011, 85, 3986–3995. [Google Scholar] [CrossRef]
- Schliephake, A.W.; Rethwilm, A. Nuclear localization of foamy virus Gag precursor protein. J. Virol. 1994, 68, 4946–4954. [Google Scholar]
- Müllers, E.; Stirnnagel, K.; Kaulfuss, S.; Lindemann, D. Prototype foamy virus gag nuclear localization: A novel pathway among retroviruses. J. Virol. 2011, 85, 9276–9285. [Google Scholar] [CrossRef]
- Heinkelein, M.; Pietschmann, T.; Jarmy, G.; Dressler, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Bock, M.; Moebes, A.; Roy, J.; et al. Efficient intracellular retrotransposition of an exogenous primate retrovirus genome. EMBO J. 2000, 19, 3436–3445. [Google Scholar] [CrossRef]
- Heinkelein, M.; Rammling, M.; Juretzek, T.; Lindemann, D.; Rethwilm, A. Retrotransposition and cell-to-cell transfer of foamy viruses. J. Virol. 2003, 77, 11855–11858. [Google Scholar] [CrossRef]
- Bodem, J.; Zemba, M.; Flügel, R.M. Nuclear localization of the functional Bel 1 transactivator but not of the gag proteins of the feline foamy virus. Virology 1998, 251, 22–27. [Google Scholar] [CrossRef]
- Pitha, P.M. Innate antiviral response: Role in HIV-1 infection. Viruses 2011, 3, 1179–1203. [Google Scholar] [CrossRef]
- Meyerson, N.R.; Sawyer, S.L. Two-stepping through time: Mammals and viruses. Trends Microbiol. 2011, 19, 286–294. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Venkatesh, S.; Bieniasz, P.D. Intrinsic cellular defenses against human immunodeficiency viruses. Immunity 2012, 37, 399–411. [Google Scholar] [CrossRef]
- Rhodes-Feuillette, A.; Lasneret, J.; Paulien, S.; Ogunkolade, W.; Peries, J.; Canivet, M. Effects of human recombinant alpha and gamma and of highly purified natural beta interferons on simian Spumavirinae prototype (simian foamy virus 1) multiplication in human cells. Res. Virol. 1990, 141, 31–43. [Google Scholar] [CrossRef]
- Rhodes-Feuillette, A.; Saal, F.; Lasneret, J.; Santillana-Hayat, M.; Peries, J. Studies on in vitro interferon induction capacity and interferon sensivity of simian foamy viruses. Arch. Virol. 1987, 97, 77–84. [Google Scholar] [CrossRef]
- Falcone, V.; Schweizer, M.; Toniolo, A.; Neumann-Haefelin, D.; Meyerhans, A. Gamma interferon is a major suppressive factor produced by activated human peripheral blood lymphocytes that is able to inhibit foamy virus-induced cytopathic effects. J. Virol. 1999, 73, 1724–1728. [Google Scholar]
- Von Laer, D.; Neumann-Haefelin, D.; Heeney, J.L.; Schweizer, M. Lymphocytes are the major reservoir for foamy viruses in peripheral blood. Virology 1996, 221, 240–244. [Google Scholar] [CrossRef]
- Liberatore, R.A.; Bieniasz, P.D. Sensing retroviruses. Immunity 2011, 35, 8–10. [Google Scholar] [CrossRef]
- Kane, M.; Case, L.K.; Wang, C.; Yurkovetskiy, L.; Dikiy, S.; Golovkina, T.V. Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 2011, 35, 135–145. [Google Scholar] [CrossRef]
- Rua, R.; Lepelley, A.; Gessain, A.; Schwartz, O. Innate sensing of foamy viruses by human hematopoietic cells. J. Virol. 2012, 86, 909–918. [Google Scholar] [CrossRef]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Yu, S.F.; Sullivan, M.D.; Linial, M.L. Evidence that the human foamy virus genome is DNA. J. Virol. 1999, 73, 1565–1572. [Google Scholar]
- Wolf, D.; Goff, S.P. Host restriction factors blocking retroviral replication. Annu. Rev. Genet. 2008, 42, 143–163. [Google Scholar] [CrossRef]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Jeang, K.T.; Tokunaga, K. Host restriction factors in retroviral infection: Promises in virus-host interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef]
- Callahan, M.E.; Switzer, W.M.; Matthews, A.L.; Roberts, B.D.; Heneine, W.; Folks, T.M.; Sandstrom, P.A. Persistent zoonotic infection of a human with simian foamy virus in the absence of an intact orf-2 accessory gene. J. Virol. 1999, 73, 9619–9624. [Google Scholar]
- Nisole, S.; Stoye, J.P.; Saib, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef]
- Towers, G.J. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 2007, 4, 40. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef]
- Black, L.R.; Aiken, C. TRIM5alpha disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef]
- Lukic, Z.; Hausmann, S.; Sebastian, S.; Rucci, J.; Sastri, J.; Robia, S.L.; Luban, J.; Campbell, E.M. TRIM5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. Retrovirology 2011, 8, 93. [Google Scholar] [CrossRef]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Yang, A.; Cowan, S.; Bieniasz, P.D. Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J. Virol. 2005, 79, 8969–8978. [Google Scholar] [CrossRef]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol.: CB 2005, 15, 73–78. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Flower, T.G.; Ball, N.J.; Sanz-Ramos, M.; Yap, M.W.; Ogrodowicz, R.W.; Stanke, N.; Reh, J.; Lindemann, D.; Stoye, J.P.; et al. A unique spumavirus Gag N-terminal domain with functional properties of orthoretroviral matrix and capsid. PLoS Pathog. 2013, 9, e1003376. [Google Scholar] [CrossRef]
- Yap, M.W.; Lindemann, D.; Stanke, N.; Reh, J.; Westphal, D.; Hanenberg, H.; Ohkura, S.; Stoye, J.P. Restriction of foamy viruses by primate Trim5alpha. J. Virol. 2008, 82, 5429–5439. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Princiotta, M.; Piatak, M., Jr.; Yuan, F.; Zhang, F.; Lifson, J.D.; Bieniasz, P.D. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 2006, 314, 95. [Google Scholar] [CrossRef]
- Pacheco, B.; Finzi, A.; McGee-Estrada, K.; Sodroski, J. Species-specific inhibition of foamy viruses from South American monkeys by New World Monkey TRIM5{alpha} proteins. J. Virol. 2010, 84, 4095–4099. [Google Scholar] [CrossRef]
- Cullen, B.R. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 2006, 80, 1067–1076. [Google Scholar] [CrossRef]
- Malim, M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 675–687. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: Not simply editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol.: CB 2005, 15, 166–170. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Jaguva Vasudevan, A.A.; Perkovic, M.; Bulliard, Y.; Cichutek, K.; Trono, D.; Haussinger, D.; Munk, C. Prototype Foamy Virus Bet Impairs the Dimerization and Cytosolic Solubility of Human APOBEC3G. J. Virol. 2013. [Google Scholar] [CrossRef]
- Slavkovic Lukic, D.; Hotz-Wagenblatt, A.; Lei, J.; Räthe, A.M.; Mühle, M.; Denner, J.; Münk, C.; Löchelt, M. Identification of the feline foamy virus Bet domain essential for APOBEC3 counteraction. Retrovirology 2013, 10, 76. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef]
- Wood, N.; Bhattacharya, T.; Keele, B.F.; Giorgi, E.; Liu, M.; Gaschen, B.; Daniels, M.; Ferrari, G.; Haynes, B.F.; McMichael, A.; et al. HIV evolution in early infection: Selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 2009, 5, e1000414. [Google Scholar] [CrossRef] [Green Version]
- Perez-Caballero, D.; Soll, S.J.; Bieniasz, P.D. Evidence for restriction of ancient primate gammaretroviruses by APOBEC3 but not TRIM5alpha proteins. PLoS Pathog. 2008, 4, e1000181. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Hammonds, J.; Wang, J.J.; Spearman, P. Restriction of retroviral replication by Tetherin/BST-2. Mol. Biol. Int. 2012, 2012, 424768. [Google Scholar]
- Neil, S.J.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Xu, F.; Tan, J.; Liu, R.; Xu, D.; Li, Y.; Geng, Y.; Liang, C.; Qiao, W. Tetherin inhibits prototypic foamy virus release. Virol. J. 2011, 8, 198–207. [Google Scholar] [CrossRef]
- Dietrich, I.; McMonagle, E.L.; Petit, S.J.; Vijayakrishnan, S.; Logan, N.; Chan, C.N.; Towers, G.J.; Hosie, M.J.; Willett, B.J. Feline tetherin efficiently restricts release of feline immunodeficiency virus but not spreading of infection. J. Virol. 2011, 85, 5840–5852. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Mullers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of diverse retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef]
- Loh, P.C.; Matsuura, F.; Mizumoto, C. Seroepidemiology of human syncytial virus: Antibody prevalence in the Pacific. Intervirology 1980, 13, 87–90. [Google Scholar] [CrossRef]
- Mahnke, C.; Kashaiya, P.; Rössler, J.; Bannert, H.; Levin, A.; Blattner, W.A.; Dietrich, M.; Luande, J.; Löchelt, M.; Friedman-Kien, A.E.; et al. Human spumavirus antibodies in sera from African patients. Arch. Virol. 1992, 123, 243–253. [Google Scholar] [CrossRef]
- Schweizer, M.; Turek, R.; Hahn, H.; Schliephake, A.; Netzer, K.O.; Eder, G.; Reinhardt, M.; Rethwilm, A.; Neumann-Haefelin, D. Markers of foamy virus infections in monkeys, apes, and accidentally infected humans: Appropriate testing fails to confirm suspected foamy virus prevalence in humans. AIDS Res. Hum. Retrovir. 1995, 11, 161–170. [Google Scholar] [CrossRef]
- Ali, M.; Taylor, G.P.; Pitman, R.J.; Parker, D.; Rethwilm, A.; Cheingsong-Popov, R.; Weber, J.N.; Bieniasz, P.D.; Bradley, J.; McClure, M.O. No evidence of antibody to human foamy virus in widespread human populations. AIDS Res. Hum. Retrovir. 1996, 12, 1473–1483. [Google Scholar] [CrossRef]
- Achong, B.G.; Mansell, P.W.; Epstein, M.A.; Clifford, P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971, 46, 299–307. [Google Scholar]
- Herchenröder, O.; Renne, R.; Loncar, D.; Cobb, E.K.; Murthy, K.K.; Schneider, J.; Mergia, A.; Luciw, P.A. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): High homology to human foamy virus (HFV). Virology 1994, 201, 187–199. [Google Scholar] [CrossRef]
- Jones-Engel, L.; May, C.C.; Engel, G.A.; Steinkraus, K.A.; Schillaci, M.A.; Fuentes, A.; Rompis, A.; Chalise, M.K.; Aggimarangsee, N.; Feeroz, M.M.; et al. Diverse contexts of zoonotic transmission of simian foamy viruses in Asia. Emerg. Infect. Dis. 2008, 14, 1200–1208. [Google Scholar] [CrossRef]
- Keele, B.F.; Jones, J.H.; Terio, K.A.; Estes, J.D.; Rudicell, R.S.; Wilson, M.L.; Li, Y.; Learn, G.H.; Beasley, T.M.; Schumacher-Stankey, J.; et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 2009, 460, 515–519. [Google Scholar] [CrossRef]
- Huang, F.; Yu, W. Foamy virus in the tree shrew Tupaia belangeri is highly related to simian foamy virus in Macaca Mulatta. AIDS Res. Hum. Retrovir. 2013, 29, 1177–1178. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. An endogenous foamy virus in the aye-aye (Daubentonia madagascariensis). J. Virol. 2012, 86, 7696–7698. [Google Scholar] [CrossRef]
- Saib, A. Non-primate foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 197–211. [Google Scholar] [CrossRef]
- Meiering, C.D.; Linial, M.L. Historical perspective of foamy virus epidemiology and infection. Clin. Microbiol. Rev. 2001, 14, 165–176. [Google Scholar] [CrossRef]
- Flanagan, M. Isolation of a spumavirus from a sheep. Aust. Vet. J. 1992, 69, 112–113. [Google Scholar] [CrossRef]
- Amborski, G.F.; Storz, J.; Keney, D.; Lo, J.; McChesney, A.E. Isolation of a retrovirus from the American bison and its relation to bovine retroviruses. J. Wildl. Dis. 1987, 23, 7–11. [Google Scholar]
- Kennedy-Stoskopf, S.; Stoskopf, M.K.; Eckhaus, M.A.; Strandberg, J.D. Isolation of a retrovirus and a herpesvirus from a captive California sea lion. J. Wildl. Dis. 1986, 22, 156–164. [Google Scholar]
- Luis, A.D.; Hayman, D.T.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.; Mills, J.N.; Timonin, M.E.; Willis, C.K.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. R. Soc. 2013, 280, 20122753. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar]
- Drexler, J.F.; Seelen, A.; Corman, V.M.; Fumie Tateno, A.; Cottontail, V.; Melim Zerbinati, R.; Gloza-Rausch, F.; Klose, S.M.; Adu-Sarkodie, Y.; Oppong, S.K.; et al. Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae. J. Virol. 2012, 86, 9134–9147. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Holmes, E.C. The evolution of endogenous viral elements. Cell Host Microbe 2011, 10, 368–377. [Google Scholar] [CrossRef]
- Cordonnier, A.; Casella, J.F.; Heidmann, T. Isolation of novel human endogenous retrovirus-like elements with foamy virus-related pol sequence. J. Virol. 1995, 69, 5890–5897. [Google Scholar]
- Benit, L.; Lallemand, J.B.; Casella, J.F.; Philippe, H.; Heidmann, T. ERV-L elements: A family of endogenous retrovirus-like elements active throughout the evolution of mammals. J. Virol. 1999, 73, 3301–3308. [Google Scholar]
- Katzourakis, A.; Gifford, R.J.; Tristem, M.; Gilbert, M.T.; Pybus, O.G. Macroevolution of complex retroviruses. Science 2009, 325, 1512. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog. 2012, 8, e1002790. [Google Scholar] [CrossRef]
- Gifford, R.J.; Katzourakis, A.; Tristem, M.; Pybus, O.G.; Winters, M.; Shafer, R.W. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 20362–20367. [Google Scholar] [CrossRef]
- Schartl, M.; Walter, R.B.; Shen, Y.; Garcia, T.; Catchen, J.; Amores, A.; Braasch, I.; Chalopin, D.; Volff, J.N.; Lesch, K.P.; et al. The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat. Genet. 2013, 45, 567–572. [Google Scholar] [CrossRef]
- Llorens, C.; Munoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct 2009, 4, 41. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rethwilm, A.; Bodem, J. Evolution of Foamy Viruses: The Most Ancient of All Retroviruses. Viruses 2013, 5, 2349-2374. https://doi.org/10.3390/v5102349
Rethwilm A, Bodem J. Evolution of Foamy Viruses: The Most Ancient of All Retroviruses. Viruses. 2013; 5(10):2349-2374. https://doi.org/10.3390/v5102349
Chicago/Turabian StyleRethwilm, Axel, and Jochen Bodem. 2013. "Evolution of Foamy Viruses: The Most Ancient of All Retroviruses" Viruses 5, no. 10: 2349-2374. https://doi.org/10.3390/v5102349
APA StyleRethwilm, A., & Bodem, J. (2013). Evolution of Foamy Viruses: The Most Ancient of All Retroviruses. Viruses, 5(10), 2349-2374. https://doi.org/10.3390/v5102349