Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
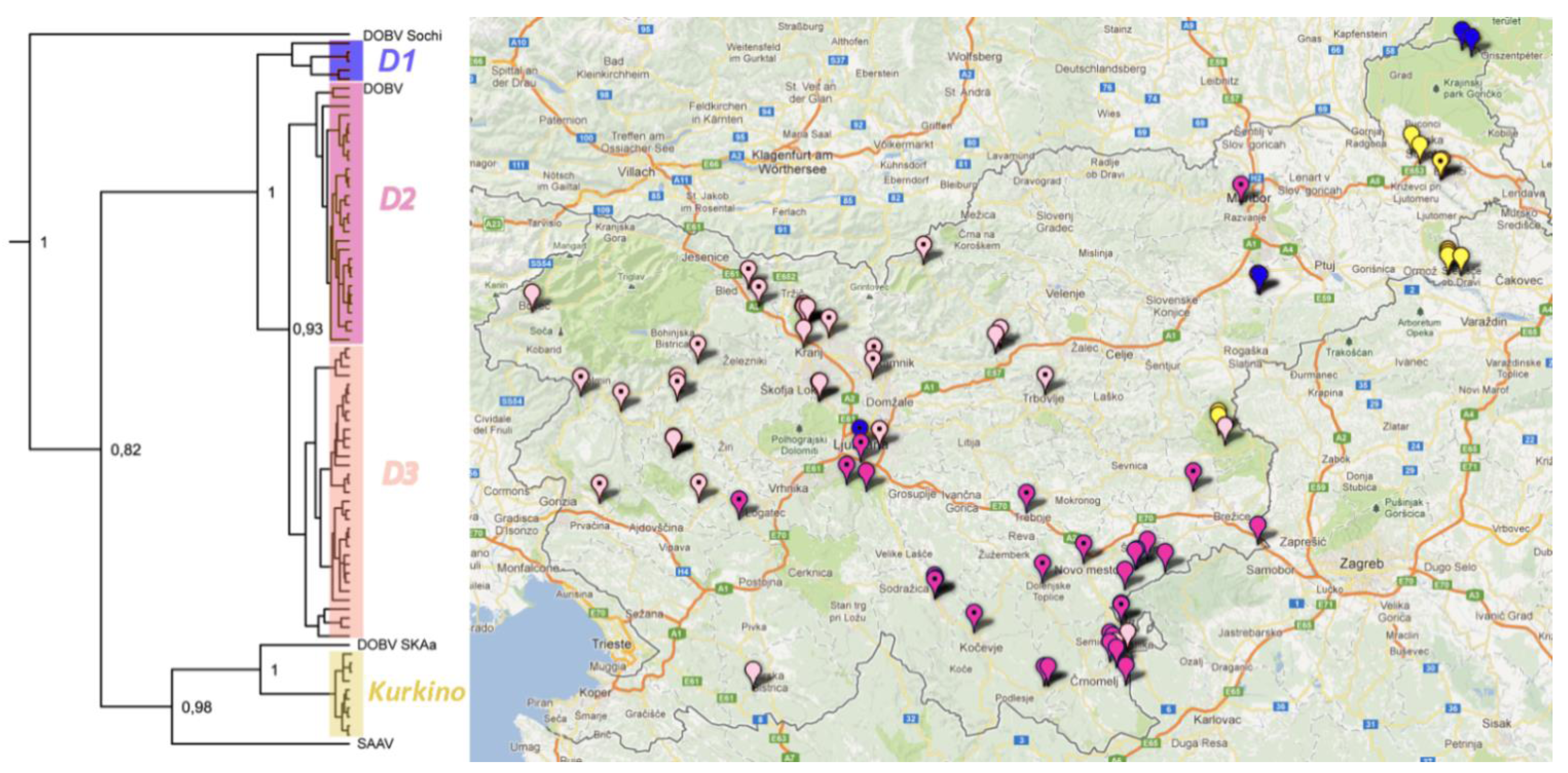
2. Results and Discussion
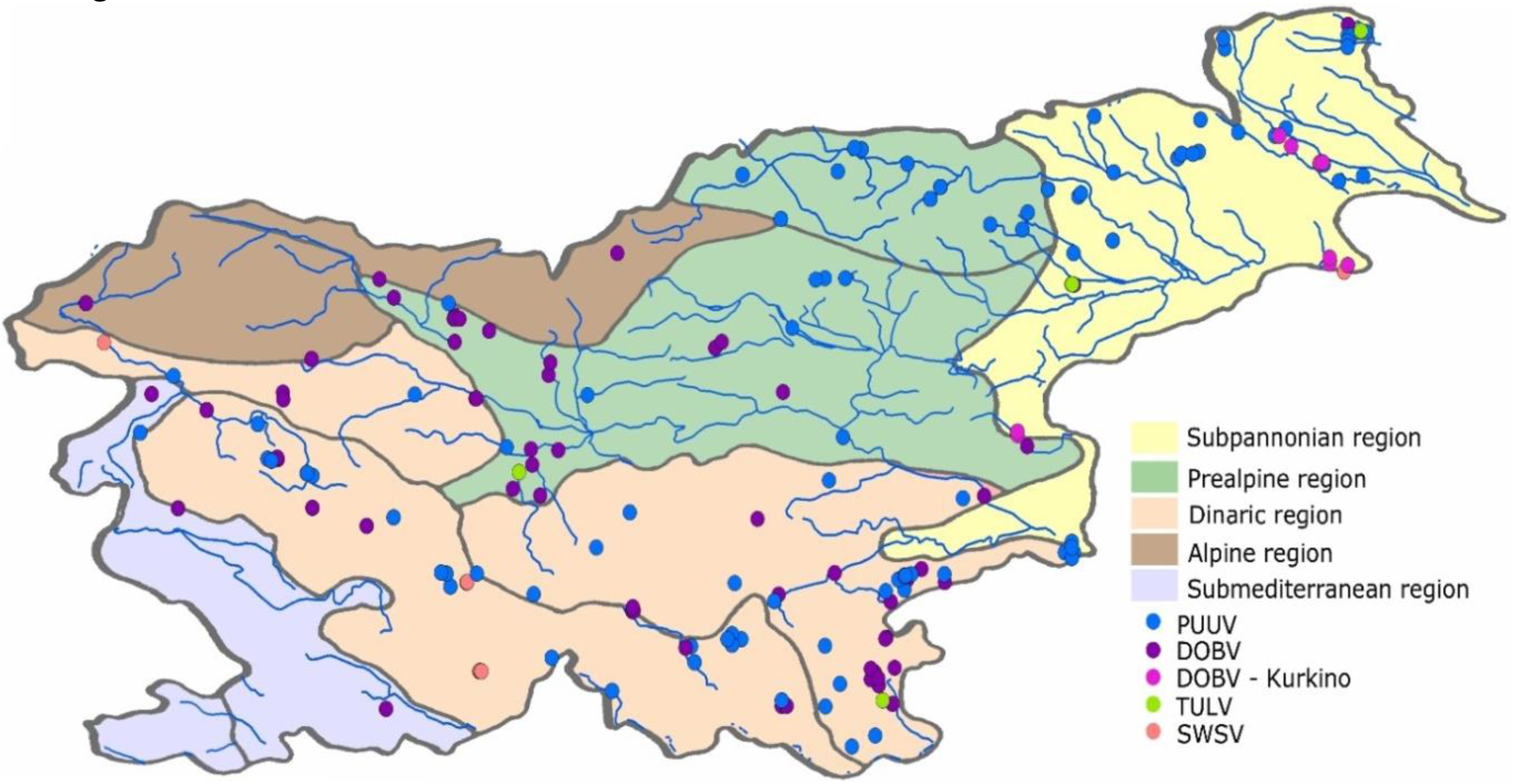
2.1. Description of Zoogeographic Regions
2.2. Collection of Animal Samples and Detection of Hantavirus RNA
2.3. Collection of Patient Samples and Detection of Hantavirus RNA

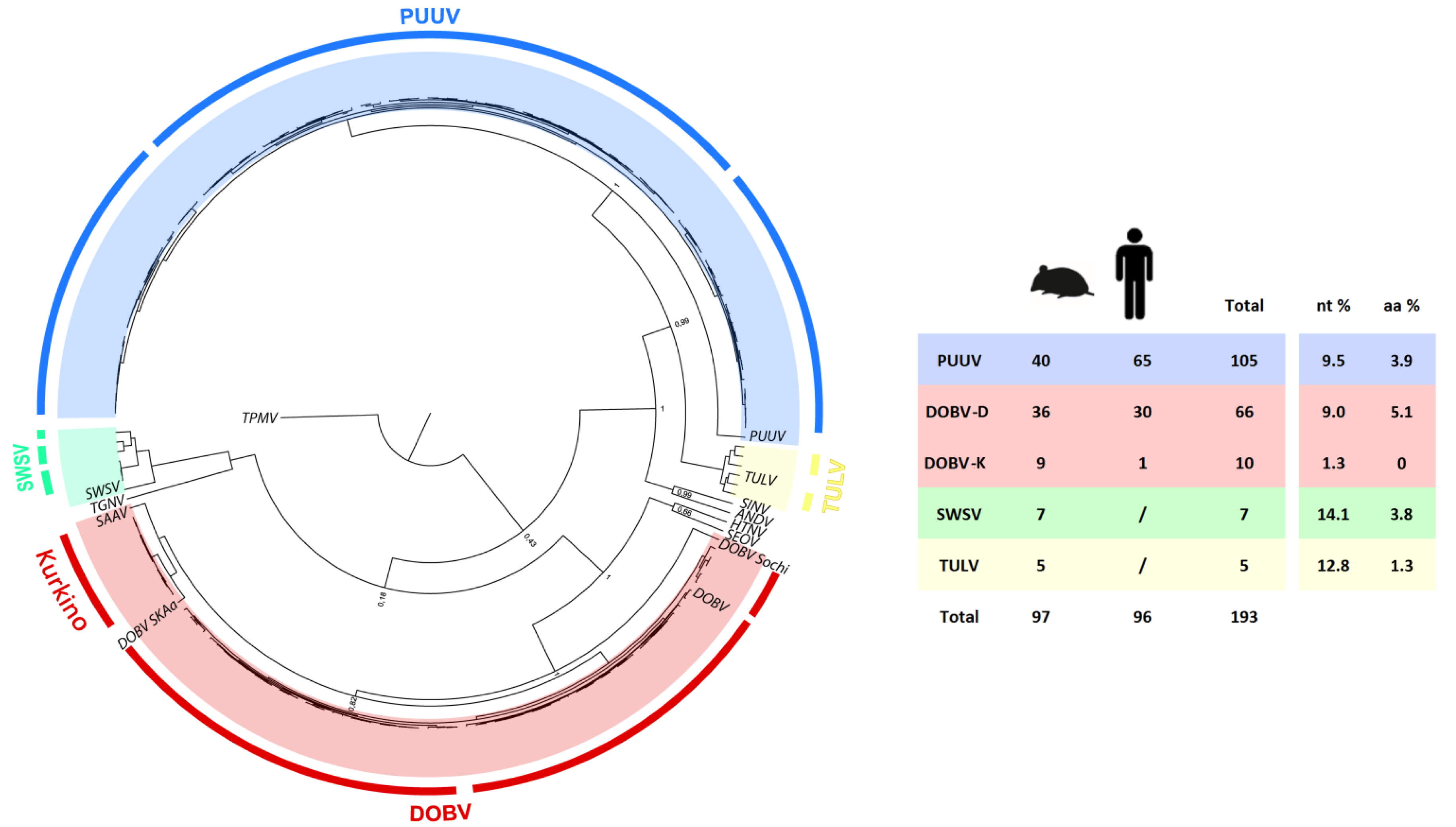
2.4. Phylogenetic Analysis L Segment Sequences

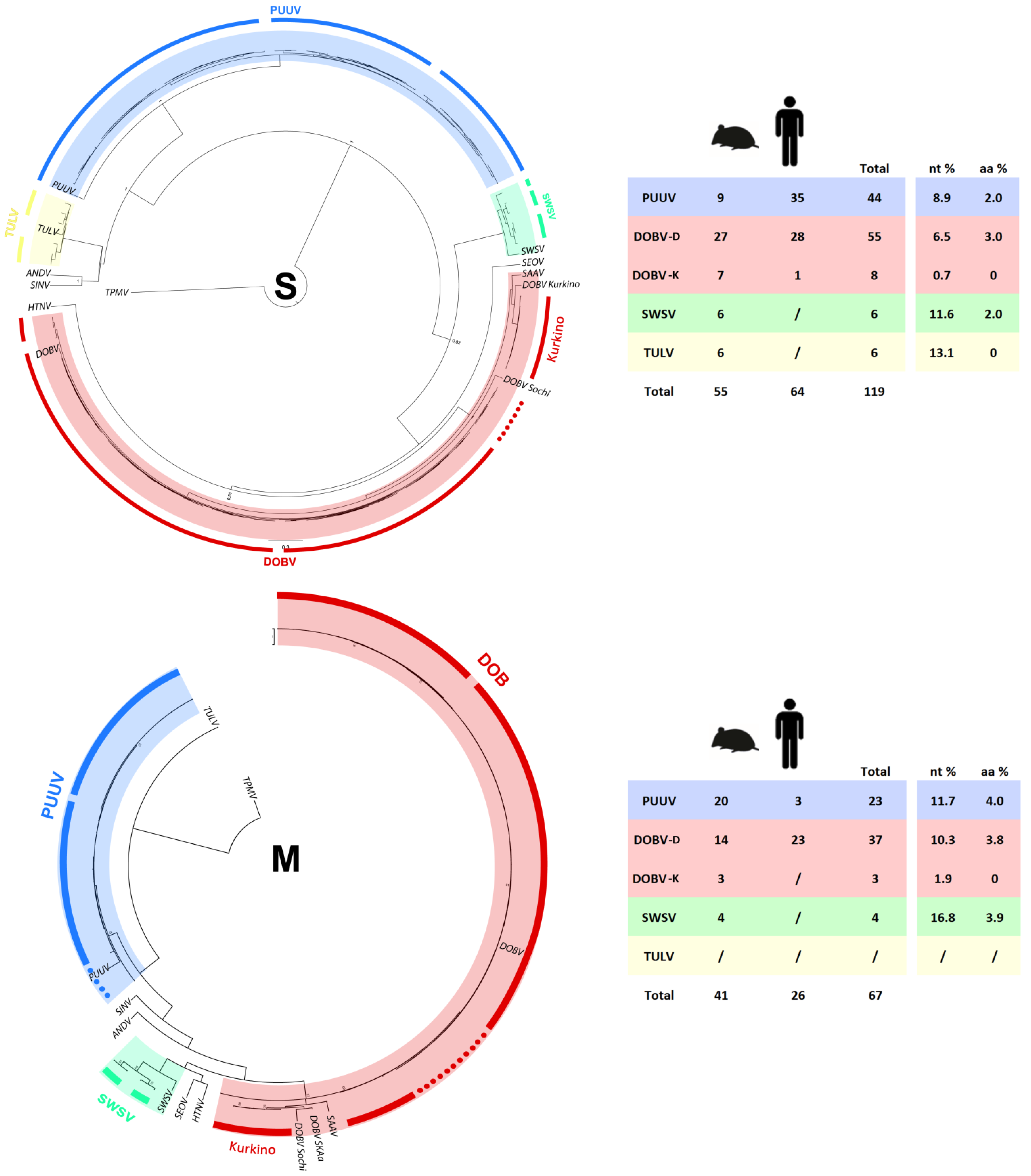
2.5. Phylogenetic Analysis of S and M Segment Sequences

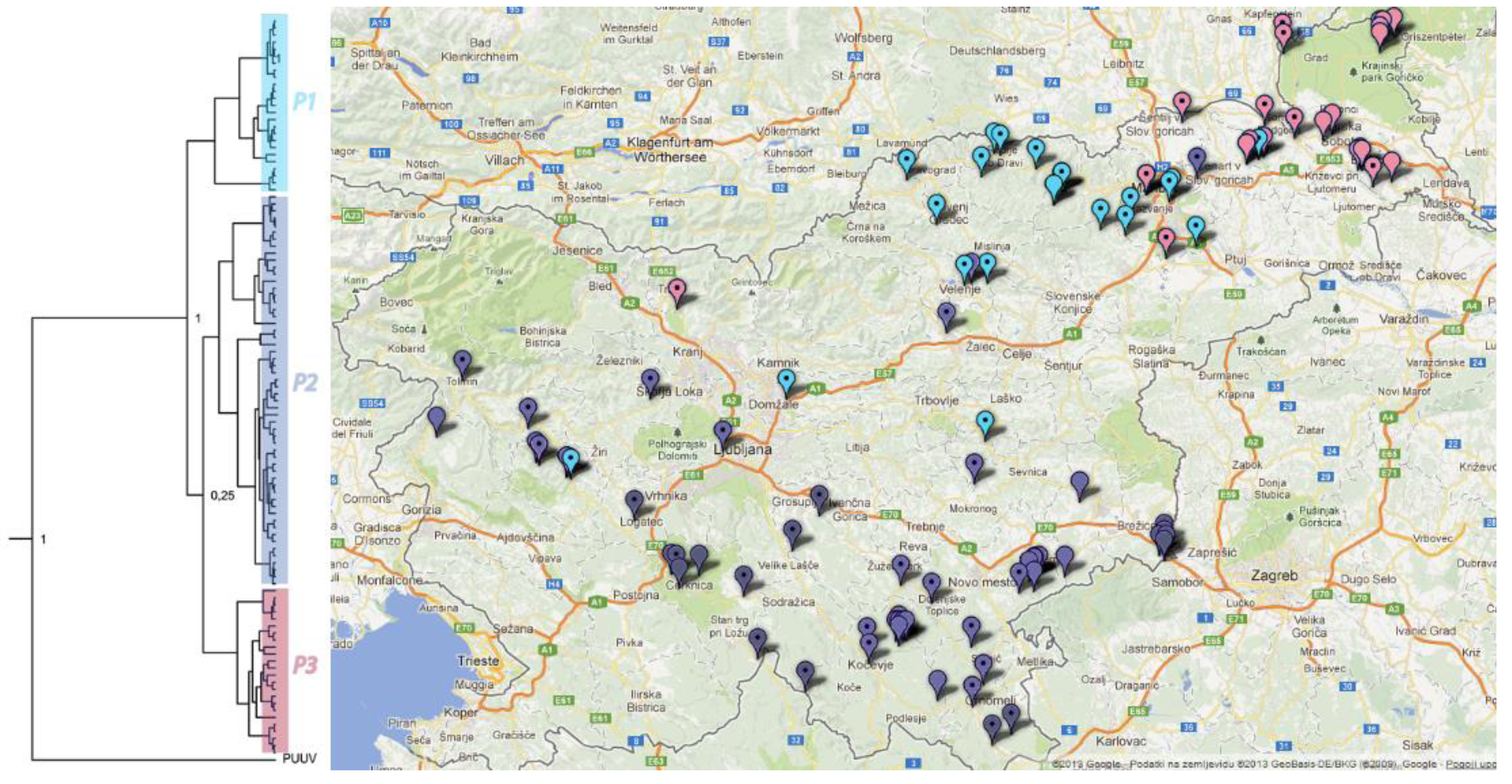
2.6. Phylogeographic Analysis


3. Experimental Section
3.1. Trapping of Small Mammals and Patient Sample Collection
3.2. DNA and RNA Extraction
3.3. Screening with Multiplex Real-Time RT-PCR for DOBV and PUUV
3.4. Screening with Hantavirus Universal RT-PCR
3.5. RT-PCR and Sequencing
3.5.1. Dobrava Virus-Specific RT-PCR
3.5.2. Dobrava-Kurkino Virus-Specific RT-PCR
3.5.3. Puumala Virus-Specific RT-PCR
3.5.4. Tula Virus-Specific RT-PCR
3.5.5. Seewis Virus-Specific RT-PCR
3.6. Phylogenetic Analysis
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Radosevic, Z.; Mohacek, I. The problem of nephropathia epidemica Myhrman-Zetterholm in relation to acute interstitial nephritis. Acta Med. Scand. 1954, 149, 221–228. [Google Scholar] [CrossRef]
- Avsic-Zupanc, T.; Xiao, S.Y.; Stojanovic, R.; Gligic, A.; van der Groen, G.; LeDuc, J.W. Characterization of dobrava virus: A hantavirus from Slovenia, Yugoslavia. J. Med. Virol. 1992, 38, 132–137. [Google Scholar] [CrossRef]
- Avsic-Zupanc, T.; Petrovec, M.; Furlan, P.; Kaps, R.; Elgh, F.; Lundkvist, A. Hemorrhagic fever with renal syndrome in the Dolenjska region of Slovenia--a 10-year survey. Clin. Infect. Dis. 1999, 28, 860–865. [Google Scholar]
- Avsic-Zupanc, T.; Nemirov, K.; Petrovec, M.; Trilar, T.; Poljak, M.; Vaheri, A.; Plyusnin, A. Genetic analysis of wild-type Dobrava hantavirus in Slovenia: Co-existence of two distinct genetic lineages within the same natural focus. J. Gen. Virol. 2000, 81, 1747–1755. [Google Scholar]
- Avsic-Zupanc, T.; Petrovec, M.; Duh, D.; Plyusnina, A.; Lundkvist, A.; Plyusnin, A. Puumala hantavirus in Slovenia: Analyses of S and M segment sequences recovered from patients and rodents. Virus Res. 2007, 123, 204–210. [Google Scholar]
- Korva, M.; Duh, D.; Puterle, A.; Trilar, T.; Zupanc, T.A. First molecular evidence of Tula hantavirus in Microtus voles in Slovenia. Virus Res. 2009, 144, 318–322. [Google Scholar] [CrossRef]
- Resman, K.; Korva, M.; Fajs, L.; Zidaric, T.; Trilar, T.; Zupanc, T.A. Molecular evidence and high genetic diversity of shrew-borne Seewis virus in Slovenia. Virus Res. 2013, 177, 113–117. [Google Scholar]
- Mršić, N. Biotska raznovrstnost v Sloveniji: Slovenija – »vroča točka« Evrope; Ministrstvo za okolje in prostor, Uprava RS za varstvo narave: Ljubljana, Slovenia, 1997; p. 129. [Google Scholar]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; ter Meulen, J.; Kruger, D.H. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar]
- Papa, A.; Johnson, A.M.; Stockton, P.C.; Bowen, M.D.; Spiropoulou, C.F.; Alexiou-Daniel, S.; Ksiazek, T.G.; Nichol, S.T.; Antoniadis, A. Retrospective serological and genetic study of the distribution of hantaviruses in Greece. J. Med. Virol. 1998, 55, 321–327. [Google Scholar] [CrossRef]
- Chu, Y.K.; Jennings, G.B.; Schmaljohn, C.S. A vaccinia virus-vectored Hantaan virus vaccine protects hamsters from challenge with Hantaan and Seoul viruses but not Puumala virus. J. Virol. 1995, 69, 6417–6423. [Google Scholar]
- Bowen, M.D.; Gelbmann, W.; Ksiazek, T.G.; Nichol, S.T.; Nowotny, N. Puumala virus and two genetic variants of Tula virus are present in Austrian rodents. J. Med. Virol. 1997, 53, 174–181. [Google Scholar] [CrossRef]
- Schlegel, M.; Radosa, L.; Rosenfeld, U.M.; Schmidt, S.; Triebenbacher, C.; Lohr, P.W.; Fuchs, D.; Heroldova, M.; Janova, E.; Stanko, M.; et al. Broad geographical distribution and high genetic diversity of shrew-borne Seewis hantavirus in Central Europe. Virus Genes 2012, 45, 48–55. [Google Scholar]
- Song, J.W.; Gu, S.H.; Bennett, S.N.; Arai, S.; Puorger, M.; Hilbe, M.; Yanagihara, R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 2007, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar]
- Delić, T.; Luštrik, R.; Kljun, F.; Potočnik, H. Species diversity and composition of small mammal communities in Goteniška gora (S Slovenia). Nat. Slov. 2012, 15, 13–26. [Google Scholar]
- Kryštufek, B. Sesalci Slovenije; Prirodoslovni muzej Slovenije: Ljubljana, Slovenia, 1991; pp. 150–159. [Google Scholar]
- Smith, M.J.; White, A.; Lambin, X.; Sherratt, J.A.; Begon, M. Delayed density-dependent season length alone can lead to rodent population cycles. Am. Nat. 2006, 167, 695–704. [Google Scholar]
- Scharninghausen, J.J.; Pfeffer, M.; Meyer, H.; Davis, D.S.; Honeycutt, R.L.; Faulde, M. Genetic evidence for Tula virus in Microtus arvalis and Microtus agrestis populations in Croatia. Vector Borne Zoonotic Dis. 2002, 2, 19–27. [Google Scholar]
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar]
- Mills, J. Biodiversity loss and emerging infectious disease: An example from the rodent-borne hemorrhagic fevers. Biodiversity 2006, 7, 9–17. [Google Scholar] [CrossRef]
- Suzán, G.; Marcé, E.; Giermakowski, J.T.; Armién, B.; Pascale, J.; Mills, J.; Ceballos, G.; Gómez, A.; Aguirre, A.A.; Salazar-Bravo, J.; et al. The effect of habitat fragmentation and species diversity loss on hantavirus prevalence in Panama. Ann. N. Y. Acad. Sci. 2008, 1149, 80–83. [Google Scholar]
- Madhav, N.K.; Wagoner, K.D.; Douglass, R.J.; Mills, J.N. Delayed density-dependent prevalence of Sin Nombre virus antibody in Montana deer mice (Peromyscus maniculatus) and implications for human disease risk. Vector Borne Zoonotic Dis. 2007, 7, 353–364. [Google Scholar]
- Linard, C.; Tersago, K.; Leirs, H.; Lambin, E.F. Environmental conditions and Puumala virus transmission in Belgium. Int. J. Health Geogr. 2007, 6, 55. [Google Scholar] [CrossRef]
- Heyman, P.; Thoma, B.R.; Marie, J.L.; Cochez, C.; Essbauer, S.S. In search for factors that drive hantavirus epidemics. Front. Physiol. 2012, 3, 237. [Google Scholar]
- Linard, C.; Lamarque, P.; Heyman, P.; Ducoffre, G.; Luyasu, V.; Tersago, K.; Vanwambeke, S.O.; Lambin, E.F. Determinants of the geographic distribution of Puumala virus and Lyme borreliosis infections in Belgium. Int. J. Health Geogr. 2007, 6, 15. [Google Scholar] [CrossRef]
- Loehman, R.A.; Elias, J.; Douglass, R.J.; Kuenzi, A.J.; Mills, J.N.; Wagoner, K. Prediction of Peromyscus maniculatus (deer mouse) population dynamics in Montana, USA, using satellite-driven vegetation productivity and weather data. J. Wildl. Dis. 2012, 48, 348–360. [Google Scholar] [CrossRef]
- Reusken, C.; de Vries, A.; Adema, J.; Vos, W.; Joke van der, G.; Bekker, D.; Heyman, P. First genetic detection of Tula hantavirus in wild rodents in the Netherlands. J. Infect. 2008, 57, 500–503. [Google Scholar] [CrossRef]
- Verhagen, R.; Leirs, H.; Tkachenko, E.; van der Groen, G. Ecological and epidemiological data on Hantavirus in bank vole populations in Belgium. Arch. Virol. 1986, 91, 193–205. [Google Scholar] [CrossRef]
- Goodin, D.G.; Koch, D.E.; Owen, R.D.; Chu, Y.K.; Hutchinson, J.M.S.; Jonsson, C.B. Land cover associated with hantavirus presence in Paraguay. Glob. Ecol. Biogeogr. 2006, 15, 519–527. [Google Scholar]
- Pupila, A.; Bergmanis, U. Species diversity, abundance and dynamics of small mammals in Eastern Latvia. Acta Univ. Latv. 2006, 710, 93–101. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Korva, M.; Knap, N.; Rus, K.R.; Fajs, L.; Grubelnik, G.; Bremec, M.; Knapič, T.; Trilar, T.; Županc, T.A. Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia. Viruses 2013, 5, 3071-3087. https://doi.org/10.3390/v5123071
Korva M, Knap N, Rus KR, Fajs L, Grubelnik G, Bremec M, Knapič T, Trilar T, Županc TA. Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia. Viruses. 2013; 5(12):3071-3087. https://doi.org/10.3390/v5123071
Chicago/Turabian StyleKorva, Miša, Nataša Knap, Katarina Resman Rus, Luka Fajs, Gašper Grubelnik, Matejka Bremec, Tea Knapič, Tomi Trilar, and Tatjana Avšič Županc. 2013. "Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia" Viruses 5, no. 12: 3071-3087. https://doi.org/10.3390/v5123071
APA StyleKorva, M., Knap, N., Rus, K. R., Fajs, L., Grubelnik, G., Bremec, M., Knapič, T., Trilar, T., & Županc, T. A. (2013). Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia. Viruses, 5(12), 3071-3087. https://doi.org/10.3390/v5123071