The p36 Isoform of Murine Cytomegalovirus m152 Protein Suffices for Mediating Innate and Adaptive Immune Evasion



Abstract
:1. Introduction
2. Results and Discussion
2.1. Impaired Immune Evasion Function Coincides with Quantitative Underrepresentation of Glycosylation Isoform gp48 of m152

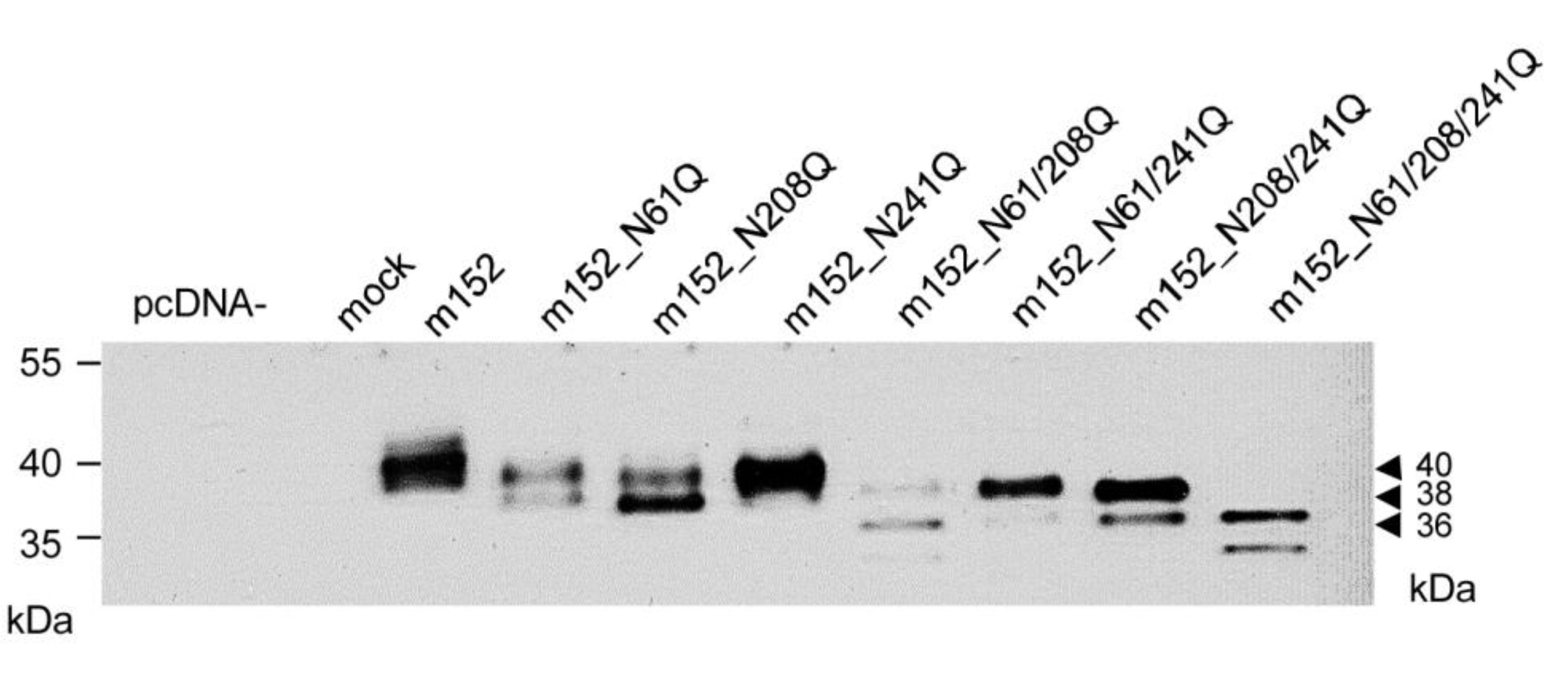
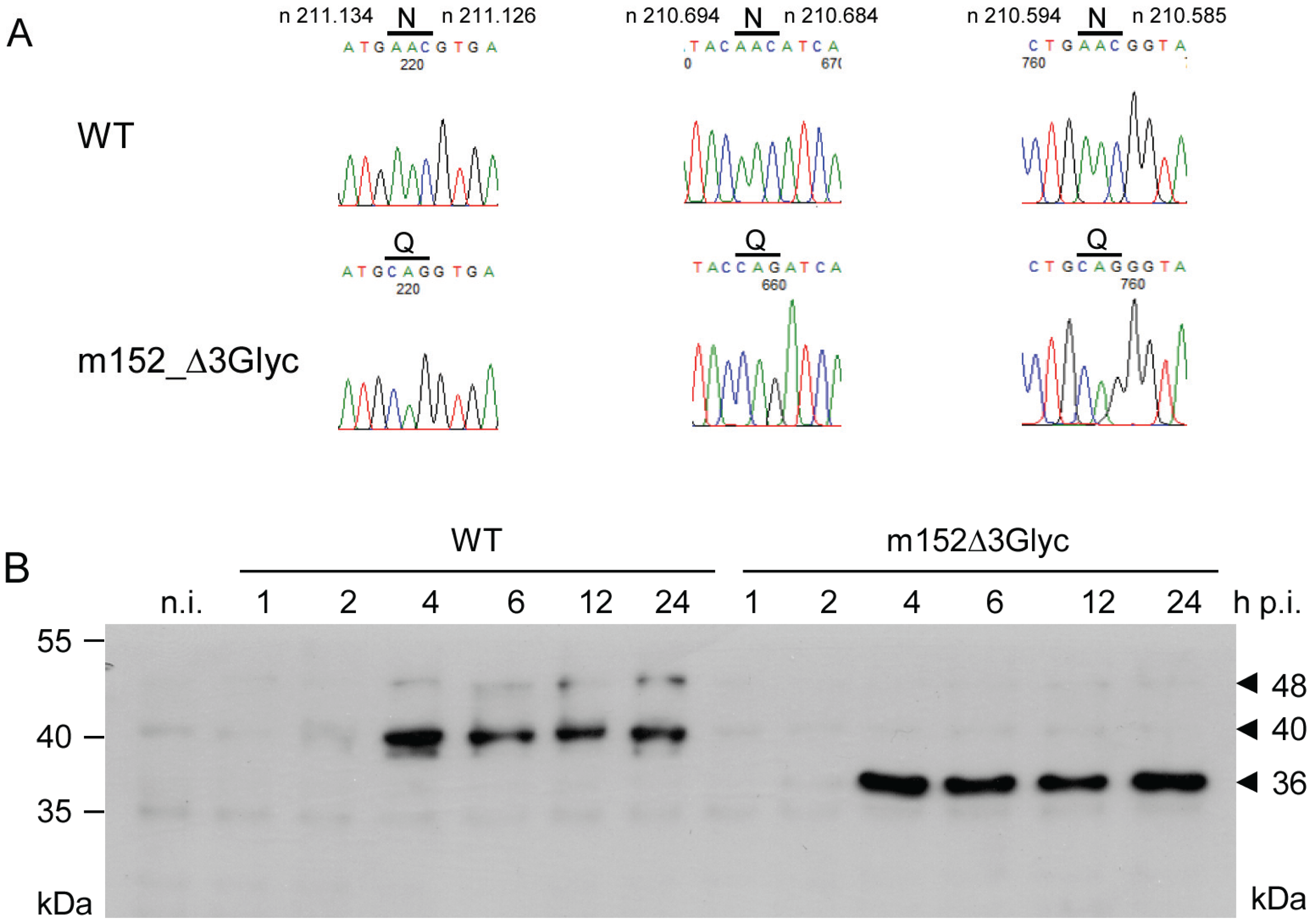
2.2. Mapping of N-glycosylation Sites in m152 and Generation of a Recombinant Virus Expressing only Isoform p36 of the m152 Gene Product


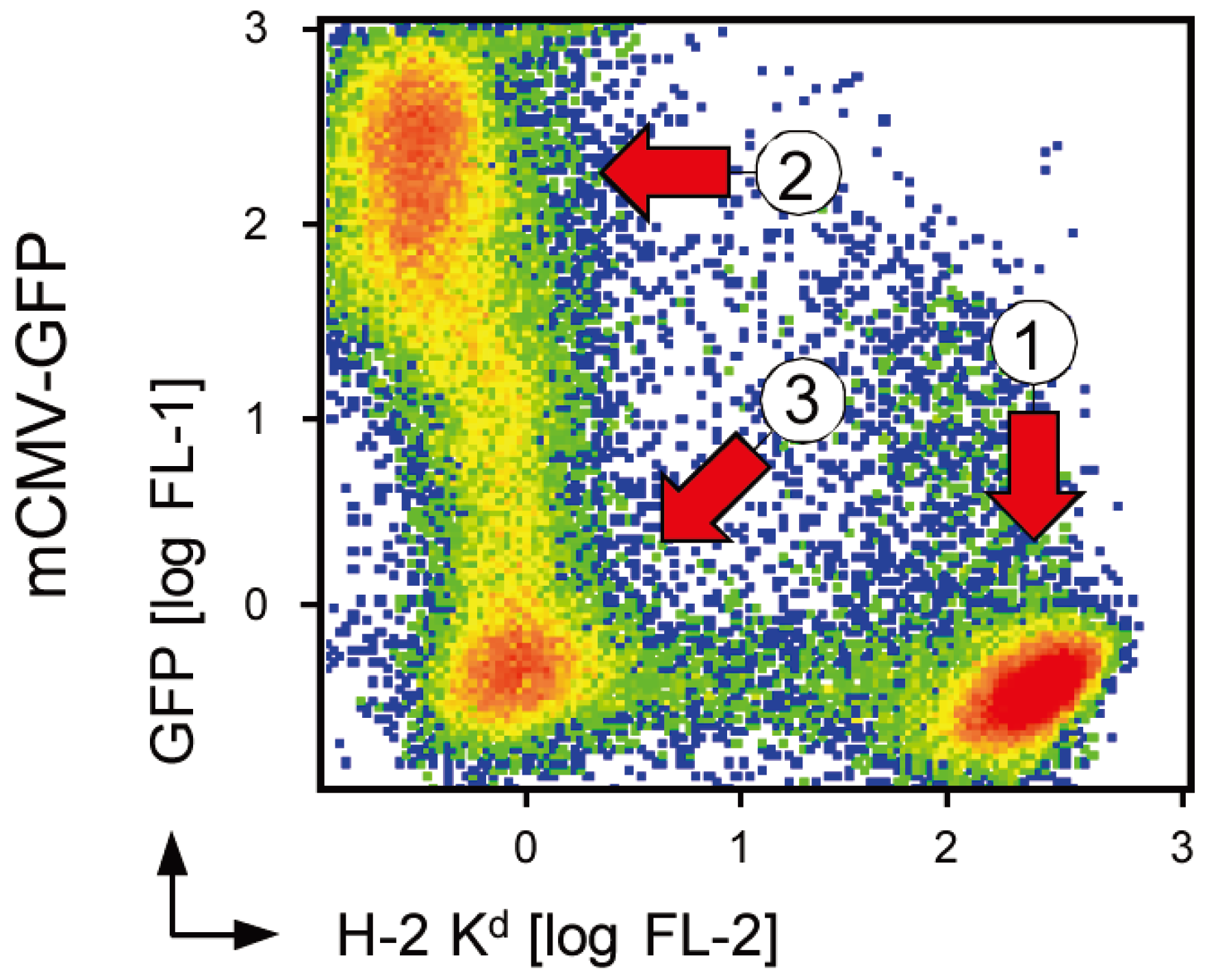
2.3. The 36 kDa Isoform of m152 Expressed by Virus mCMV-m152Δ3Glyc is Sufficient to Inhibit Antigen Presentation to CD8 T Cells
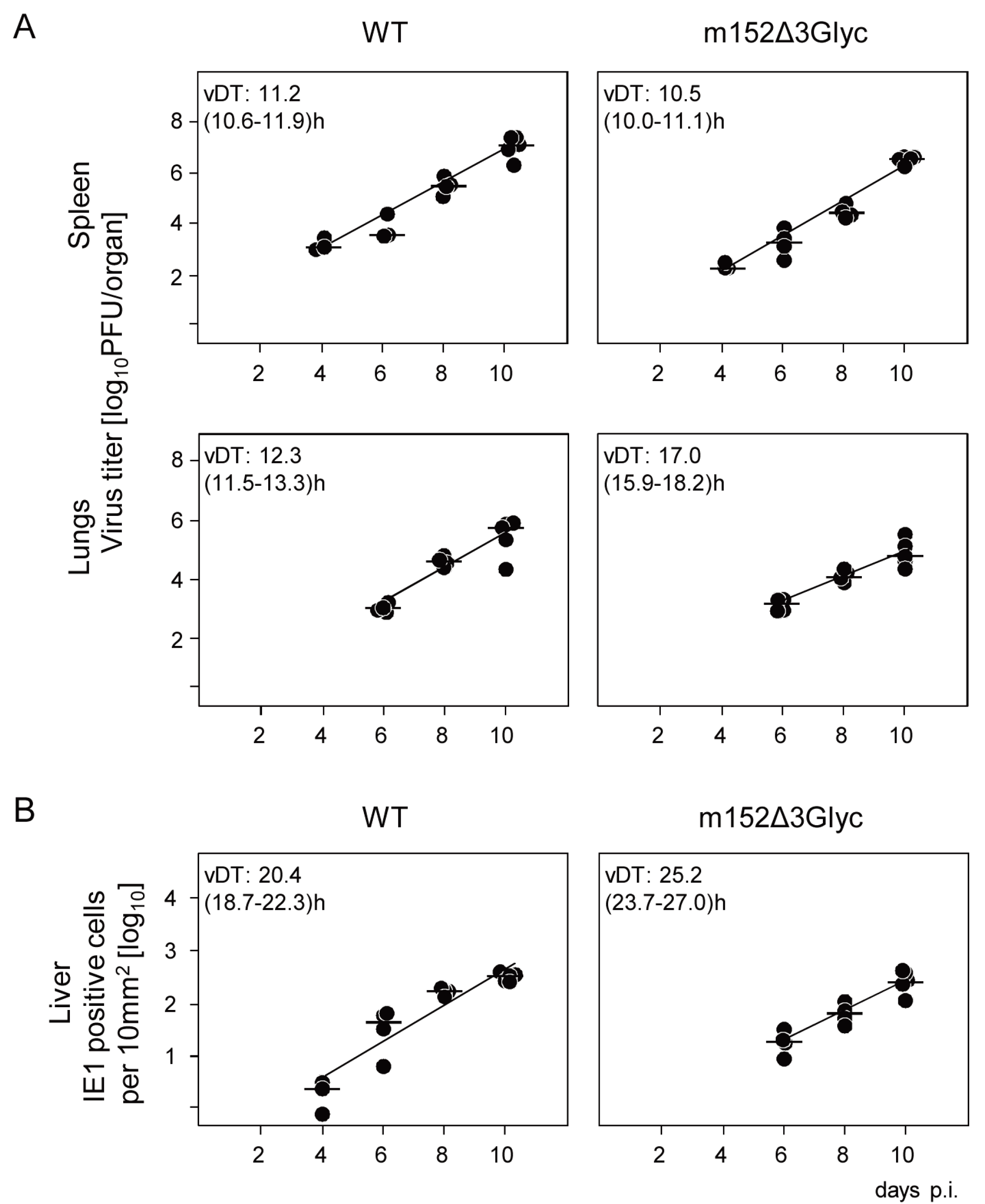
2.4. The 36 kDa Isoform of m152 Expressed by Virus mCMV-m152Δ3Glyc is Sufficient to Inhibit Activation of NK Cells through RAE1/NKG2D Interaction



3. Experimental
3.1. Cells, Viruses, and Mice
3.2. Infection Conditions and Virus Growth Kinetics in Immunocompromised Mice

3.3. In Vivo NK Cell Assays
3.4. ELISpot Analysis
3.5. Cytofluorometric Analysis
3.6. Construction of Expression Plasmids
3.7. Generation of Recombinant Viruses
3.8. Protein Extraction and Analysis
3.9. Transfection
3.10. Immunoprecipitation
3.11. Deglycosylation with Endo H and PNGase F
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Krmpotic, A.; Hasan, M.; Loewendorf, A.; Saulig, T.; Halenius, A.; Lenac, T.; Polic, B.; Bubic, I.; Kriegeskorte, A.; Pernjak-Pugel, E.; et al. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 2005, 201, 211–220. [Google Scholar] [CrossRef]
- Lodoen, M.; Ogasawara, K.; Hamerman, J.; Arase, H.; Houchins, J.; Mocarski, E.; Lanier, L. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 2003, 197, 1245–1253. [Google Scholar] [CrossRef]
- Lodoen, M.; Abenes, G.; Umamoto, S.; Houchins, J.; Liu, F.; Lanier, L. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 2004, 200, 1075–1081. [Google Scholar]
- Hasan, M.; Krmpotic, A.; Ruzsics, Z.; Bubic, I.; Lenac, T.; Halenius, A.; Loewendorf, A.; Messerle, M.; Hengel, H.; Jonjic, S.; et al. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 2005, 79, 2920–2930. [Google Scholar] [CrossRef]
- Jonjić, S.; Babić, M.; Polić, B.; Krmpotić, A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008, 20, 30–38. [Google Scholar] [CrossRef]
- Lenac, T.; Arapović, J.; Traven, L.; Krmpotić, A.; Jonjić, S. Murine cytomegalovirus regulation of NKG2D ligands. Med. Microbiol. Immunol. 2008, 197, 159–166. [Google Scholar] [CrossRef]
- Lisnić, V.J.; Krmpotić, A.; Jonjić, S. Modulation of natural killer cell activity by viruses. Curr. Opin. Microbiol. 2010, 13, 530–539. [Google Scholar] [CrossRef]
- Slavuljica, I.; Krmpotić, A.; Jonjić, S. Manipulation of NKG2D ligands by cytomegaloviruses: Impact on innate and adaptive immune response. Front. Immunol. 2011, 2. [Google Scholar] [CrossRef]
- Vidal, S.; Krmpotić, A.; Pyzik, M.; Jonjić, S. Innate immunity to cytomegalovirus in the murine model. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; pp. 191–213. [Google Scholar]
- Thäle, R.; Szepan, U.; Hengel, H.; Geginat, G.; Lucin, P.; Koszinowski, U.H. Identification of the mouse cytomegalovirus genomic region affecting major histocompatibility complex class I molecule transport. J. Virol. 1995, 69, 6098–6105. [Google Scholar]
- Del Val, M.; Hengel, H.; Häcker, H.; Hartlaub, U.; Ruppert, T.; Lucin, P.; Koszinowski, U. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J. Exp. Med. 1992, 176, 729–738. [Google Scholar] [CrossRef]
- Ziegler, H.; Thäle, R.; Lucin, P.; Muranyi, W.; Flohr, T.; Hengel, H.; Farrell, H.; Rawlinson, W.; Koszinowski, U. A mouse cytomegalovirus glycoprotein retains MHC Class I complexes in the ERGIC/ cis-Golgi compartments. Immunity 1997, 6, 57–66. [Google Scholar] [CrossRef]
- Krmpotic, A.; Messerle, M.; Crnkovic-Mertens, I.; Polic, B.; Jonjic, S.; Koszinowski, U. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 1999, 190, 1285–1296. [Google Scholar] [CrossRef]
- Krmpotić, A.; Busch, D.H.; Bubić, I.; Gebhardt, F.; Hengel, H.; Hasan, M.; Scalzo, A.A.; Koszinowski, U.H.; Jonjić, S. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 2002, 3, 529–535. [Google Scholar] [CrossRef]
- Holtappels, R.; Podlech, J.; Pahl-Seibert, M.; Jülch, M.; Thomas, D.; Simon, C.O.; Wagner, M.; Reddehase, M.J. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J. Exp. Med. 2004, 199, 131–136. [Google Scholar]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Böhm, V.; Holtappels, R.; Reddehase, M.J. In vivo impact of cytomegalovirus evasion of CD8 T-cell immunity: Facts and thoughts based on murine models. Virus Res. 2011, 157, 161–174. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Fink, A.; Podlech, J.; Ebert, S.; Wilhelmi, V.; Böhm, V.; Holtappels, R.; Reddehase, M.J. Murine cytomegalovirus immune evasion proteins operative in the MHC class I pathway of antigen processing and presentation: State of knowledge, revisions, and questions. Med. Microbiol. Immunol. 2012, 201, 497–512. [Google Scholar] [CrossRef]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Gergely, K.; Böhm, V.; Deegen, P.; Däubner, T.; Reddehase, M.J. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J. Virol. 2010, 84, 1221–1236. [Google Scholar] [CrossRef]
- Ziegler, H.; Muranyi, W.; Burgert, H.; Kremmer, E.; Koszinowski, U. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 2000, 19, 870–881. [Google Scholar] [CrossRef]
- Zhi, L.; Mans, J.; Paskow, M.J.; Brown, P.H.; Schuck, P.; Jonjić, S.; Natarajan, K.; Margulies, D.H. Direct interaction of the mouse cytomegalovirus m152/gp40 immunoevasin with RAE-1 isoforms. Biochemistry 2010, 49, 2443–2453. [Google Scholar] [CrossRef]
- Arapovic, J.; Lenac, T.; Antulov, R.; Polic, B.; Ruzsics, Z.; Carayannopoulos, L.N.; Koszinowski, U.H.; Krmpotic, A.; Jonjic, S. Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J. Virol. 2009, 83, 8198–8207. [Google Scholar] [CrossRef]
- Wang, R.; Natarajan, K.; Revilleza, M.; Boyd, L.; Zhi, L.; Zhao, H.; Robinson, H.; Margulies, D. Structural basis of mouse cytomegalovirus m152/gp40 interaction with RAE1γ reveals a paradigm for MHC/MHC interaction in immune evasion. Proc. Natl. Acad. Sci. USA 2012, 109, 3578–3587. [Google Scholar] [CrossRef]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.G.; Hengel, H.; Koszinowski, U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef]
- Fink, A.; Lemmermann, N.A.; Gillert-Marien, D.; Thomas, D.; Freitag, K.; Böhm, V.; Wilhelmi, V.; Reifenberg, K.; Reddehase, M.J.; Holtappels, R. Antigen presentation under the influence of “immune evasion” proteins and its modulation by interferon-gamma: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 513–525. [Google Scholar] [CrossRef]
- Wagner, M.; Gutermann, A.; Podlech, J.; Reddehase, M.J.; Koszinowski, U.H. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 2002, 196, 805–816. [Google Scholar] [CrossRef]
- Kleijnen, M.; Huppa, J.; Lucin, P.; Mukherjee, S.; Farrell, H.; Campbell, A.; Koszinowski, U.H.; Hill, A.; Ploegh, H. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 1997, 16, 685–694. [Google Scholar] [CrossRef]
- Däubner, T.; Fink, A.; Seitz, A.; Tenzer, S.; Müller, J.; Strand, D.; Seckert, C.K.; Janssen, C.; Renzaho, A.; Grzimek, N.K.; et al. A novel transmembrane domain mediating retention of a highly motile herpesvirus glycoprotein in the endoplasmic reticulum. J. Gen. Virol. 2010, 91, 1524–1534. [Google Scholar] [CrossRef]
- Holtappels, R.; Gillert-Marien, D.; Thomas, D.; Podlech, J.; Deegen, P.; Herter, S.; Oehrlein-Karpi, S.; Strand, D.; Wagner, M.; Reddehase, M.J. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 2006, 80, 7613–7624. [Google Scholar] [CrossRef]
- Angulo, A.; Ghazal, P.; Messerle, M. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 2000, 74, 11129–11136. [Google Scholar] [CrossRef]
- Podlech, J. Personal communication, University Medical Center Mainz: Mainz, Germany, 2013.
- Erlach, K.C.; Böhm, V.; Knabe, M.; Deegen, P.; Reddehase, M.J.; Podlech, J. Activation of hepatic natural killer cells and control of liver-adapted lymphoma in the murine model of cytomegalovirus infection. Med. Microbiol. Immunol. 2008, 197, 167–178. [Google Scholar] [CrossRef]
- Slavuljica, I.; Busche, A.; Babić, M.; Mitrović, M.; Gašparović, I.; Cekinović, D.; Markova Car, E.; Pernjak Pugel, E.; Ciković, A.; Lisnić, V.J.; et al. Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J. Clin. Invest. 2010, 120, 4532–4545. [Google Scholar] [CrossRef]
- Rawlinson, W.; Farrell, H.; Barrell, B. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar]
- Wagner, M.; Jonjic, S.; Koszinowski, U.; Messerle, M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 1999, 73, 7056–7060. [Google Scholar]
- Kurz, S.; Steffens, H.; Mayer, A.; Harris, J.; Reddehase, M. Latency versus persistence or intermittent recurrences: Evidence for a latent state of murine cytomegalovirus in the lungs. J. Virol. 1997, 71, 2980–2987. [Google Scholar]
- Lemmermann, N.A.; Podlech, J.; Seckert, C.; Kropp, K.; Grzimek, N.K.; Reddehase, M.J.; Holtappels, R. CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In Methods in Microbiology; Kabelitz, D., Kaufmann, S., Eds.; Academic Press: London, UK, 2010; pp. 369–420. [Google Scholar]
- Wolfram Mathematica, version 9, Wolfram Research: Champaign, IL, USA, 2012.
- Ho, E.L.; Carayannopoulos, L.N.; Poursine-Laurent, J.; Kinder, J.; Plougastel, B.; Smith, H.R.; Yokoyama, W.M. Costimulation of multiple NK cell activation receptors by NKG2D. J. Immunol. 2002, 169, 3667–3675. [Google Scholar]
- Böhm, V.; Simon, C.; Podlech, P.; Seckert, C.; Gendig, D.; Deegen, P.; Gillert-Marien, D.; Lemmermann, N.A.; Holtappels, R.; Reddehase, M.J. The immune evasion paradox: Immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J. Virol. 2008, 82, 11637–11650. [Google Scholar] [CrossRef]
- CXP Acquisition, version 2.2, Beckman Coulter: Indianapolis, IN, USA, 2006.
- Borst, E.; Posfai, G.; Pogoda, M.; Messerle, M. Mutagenesis of herpesvirus BACs by allele replacement. In Methods Mol. Biol., Bacterial Artificial Chromosomes; Zhao, S., Stodolsky, M., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 269–280. [Google Scholar]
- Tischer, B.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef]
- Warming, S.; Costantino, N.; Court, D.; Jenkins, N.; Copeland, G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef]
Appendices



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| m152-HpaI_for | 5'-GGAGTTAACCATATAAAAGCTGTCCCCCATGCCATTCGATCAGACGCGGGCTACTCCCGAAAGAGTAAC-3' |
| m152-HpaI-rev | 5'-GGAGTTAACTGACTAATAAGTTATCTTTATTGTACAAGTGTTGTGTGTTATCCCTGAGCCCATTTTCAG-3' |
| m152_N_83_Q_for | 5'-CATTTTCCTTGGATGCAGGTGAGCGAGCTGG-3' |
| m152_N_83_Q_rev | 5'-CCAGCTCGCTCACCTGCATCCAAGGAAAATG-3' |
| m152_N_230_Q_for | 5'-GGTTCCGTTGGCGTACCAGATCAGTCTCGCGAACGG-3' |
| m152_N_230_Q_rev | 5'-CCGTTCGCGAGACTGATCTGGTACGCCAACGGAACC-3' |
| m152_N_230_Q_for | 5'-GGTTCCGTTGGCGTACCAGATCAGTCTCGCGAACGG-3' |
| m152_N_263_Q_rev | 5'-CGCGAAGTCGGTACTACCCTGCAGGTCTCTGAGATCGAGC-3' |
| m152_BAC_for | 5'-GCGAGTTCGTCTCGAAG-3' |
| m152_BAC_rev | 5'-TAGACCGCCGACAATCAG-3' |
| pEPKan-S_m152N83Q_for | 5'-CTTTCGGATCGAAGCCTCCGGAGAGGTGAAACATTTTCCTTGGATGCAGGTGAGCGAGCTGGCGCAGCAACCAATTAACCAATTCTGATTAG-3' |
| pEPKan-S_m152N83Q_rev | 5'-ACGAAGAACGCACTCTCCTGCGCCAGCTCGCTCACCTGCATCCAAGGAAAATGTTTCACCAGGATGACGACGATAAGTAGGG-3' |
| m152_del_fwd | 5'-TGTCACCGCTCCACGTTTCACCGTCGGTCTCCCGATCGCTAGCCTGTACACAGGAACACTTAACGGCTGA-3' |
| m152_del_rev | 5'-GAGCACCCGACGATCTGACATTGTCCAGTGTGCCGGTCGCACGAACATCAAGGACGACGACGACAAGTAA-3' |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fink, A.; Renzaho, A.; Reddehase, M.J.; Lemmermann, N.A.W. The p36 Isoform of Murine Cytomegalovirus m152 Protein Suffices for Mediating Innate and Adaptive Immune Evasion. Viruses 2013, 5, 3171-3191. https://doi.org/10.3390/v5123171
Fink A, Renzaho A, Reddehase MJ, Lemmermann NAW. The p36 Isoform of Murine Cytomegalovirus m152 Protein Suffices for Mediating Innate and Adaptive Immune Evasion. Viruses. 2013; 5(12):3171-3191. https://doi.org/10.3390/v5123171
Chicago/Turabian StyleFink, Annette, Angeliqué Renzaho, Matthias J. Reddehase, and Niels A. W. Lemmermann. 2013. "The p36 Isoform of Murine Cytomegalovirus m152 Protein Suffices for Mediating Innate and Adaptive Immune Evasion" Viruses 5, no. 12: 3171-3191. https://doi.org/10.3390/v5123171
APA StyleFink, A., Renzaho, A., Reddehase, M. J., & Lemmermann, N. A. W. (2013). The p36 Isoform of Murine Cytomegalovirus m152 Protein Suffices for Mediating Innate and Adaptive Immune Evasion. Viruses, 5(12), 3171-3191. https://doi.org/10.3390/v5123171