The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results
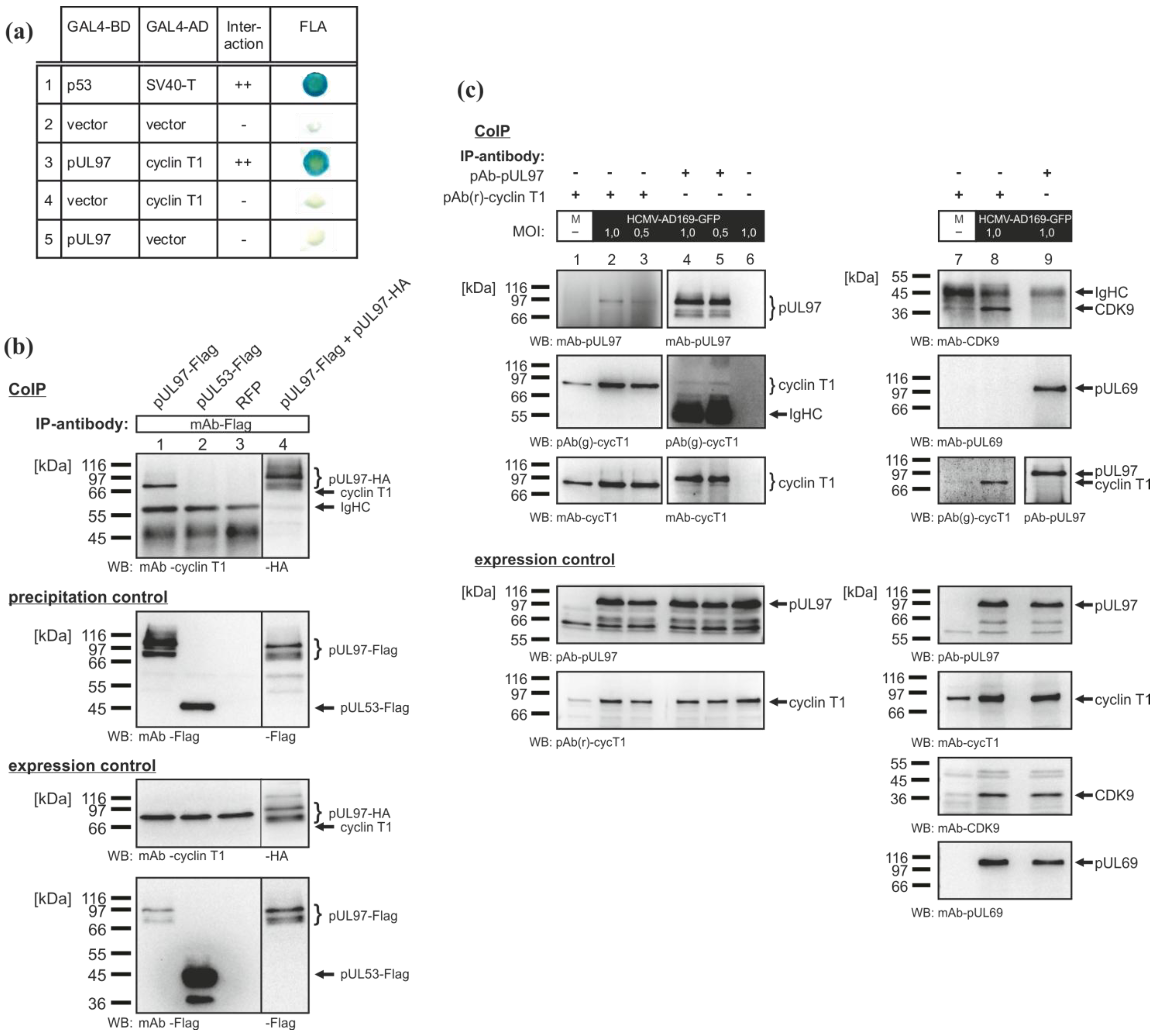
2.1. Interaction of the HCMV Protein Kinase pUL97 with Cyclin T1
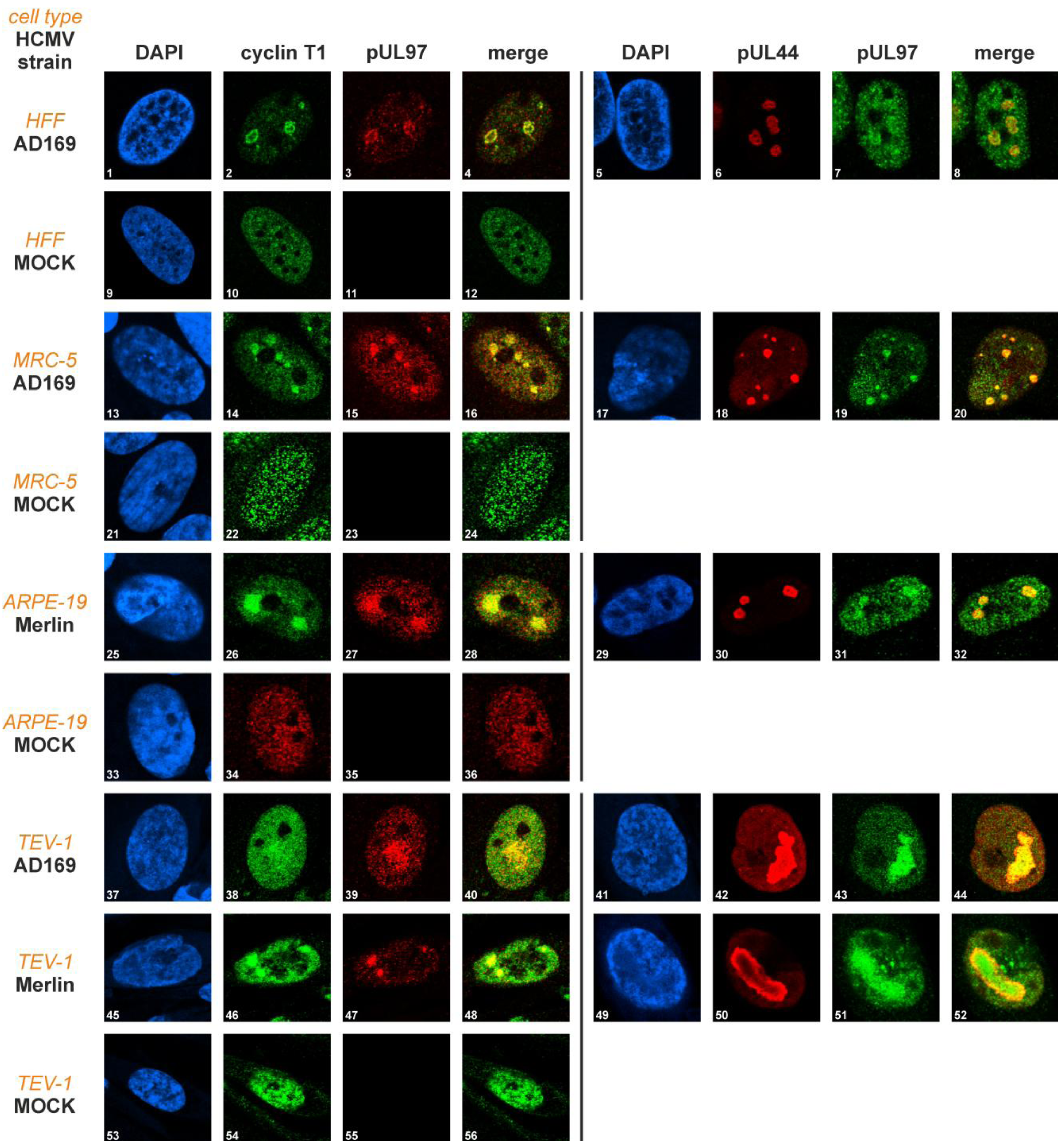
2.2. Partial Colocalization of pUL97 with Cyclin T1 in Subnuclear Compartments of HCMV-Infected Cells


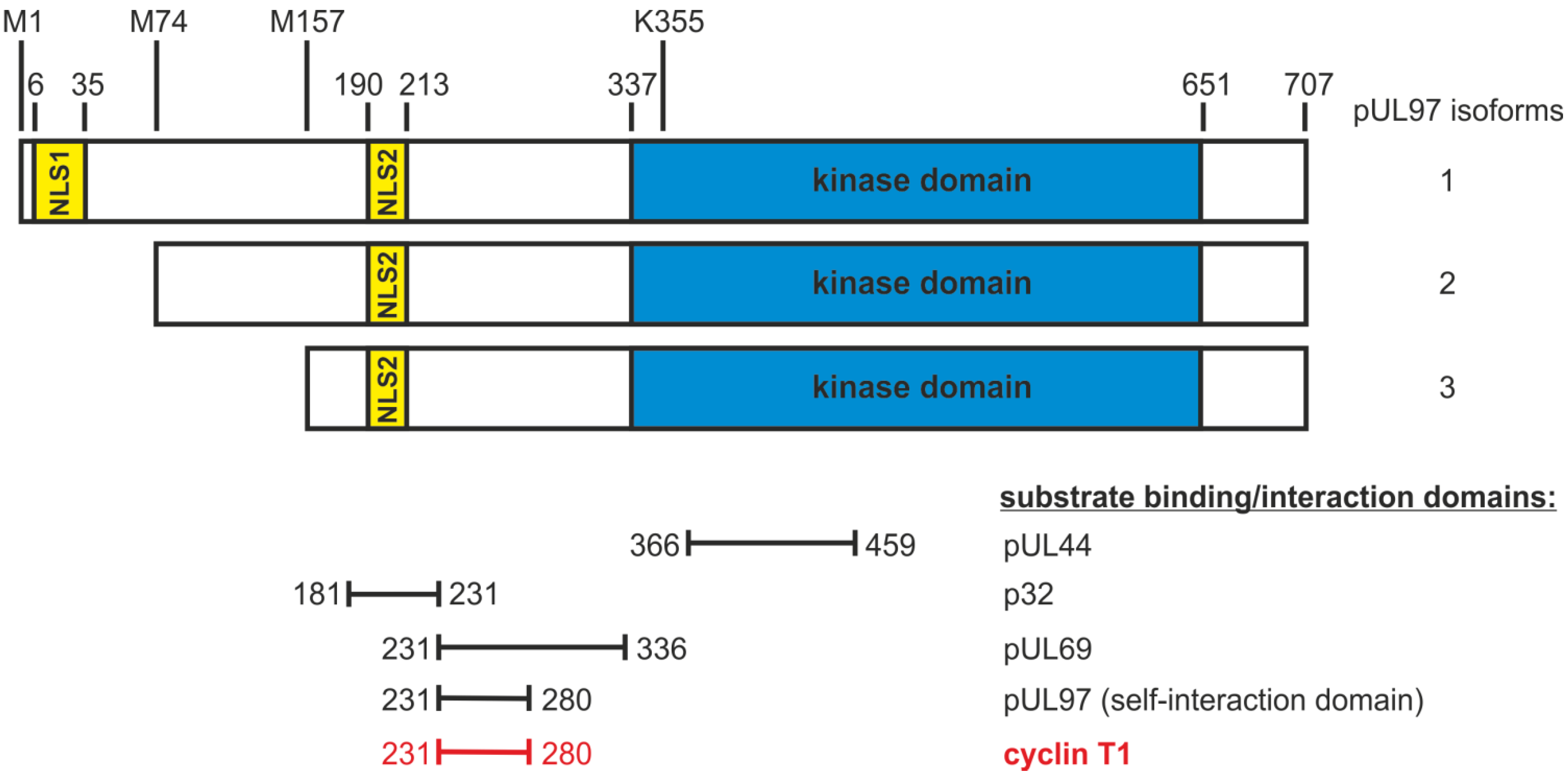
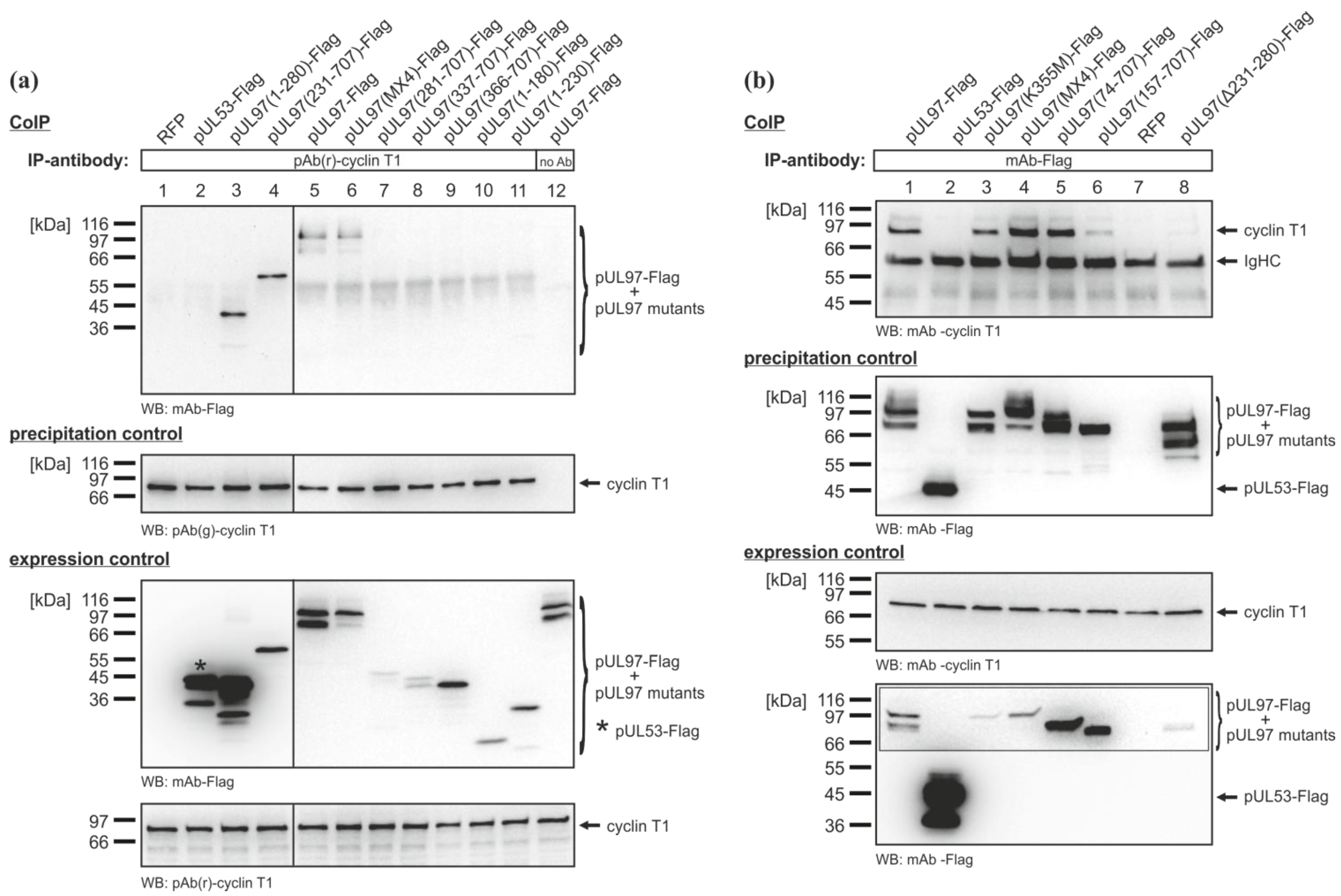
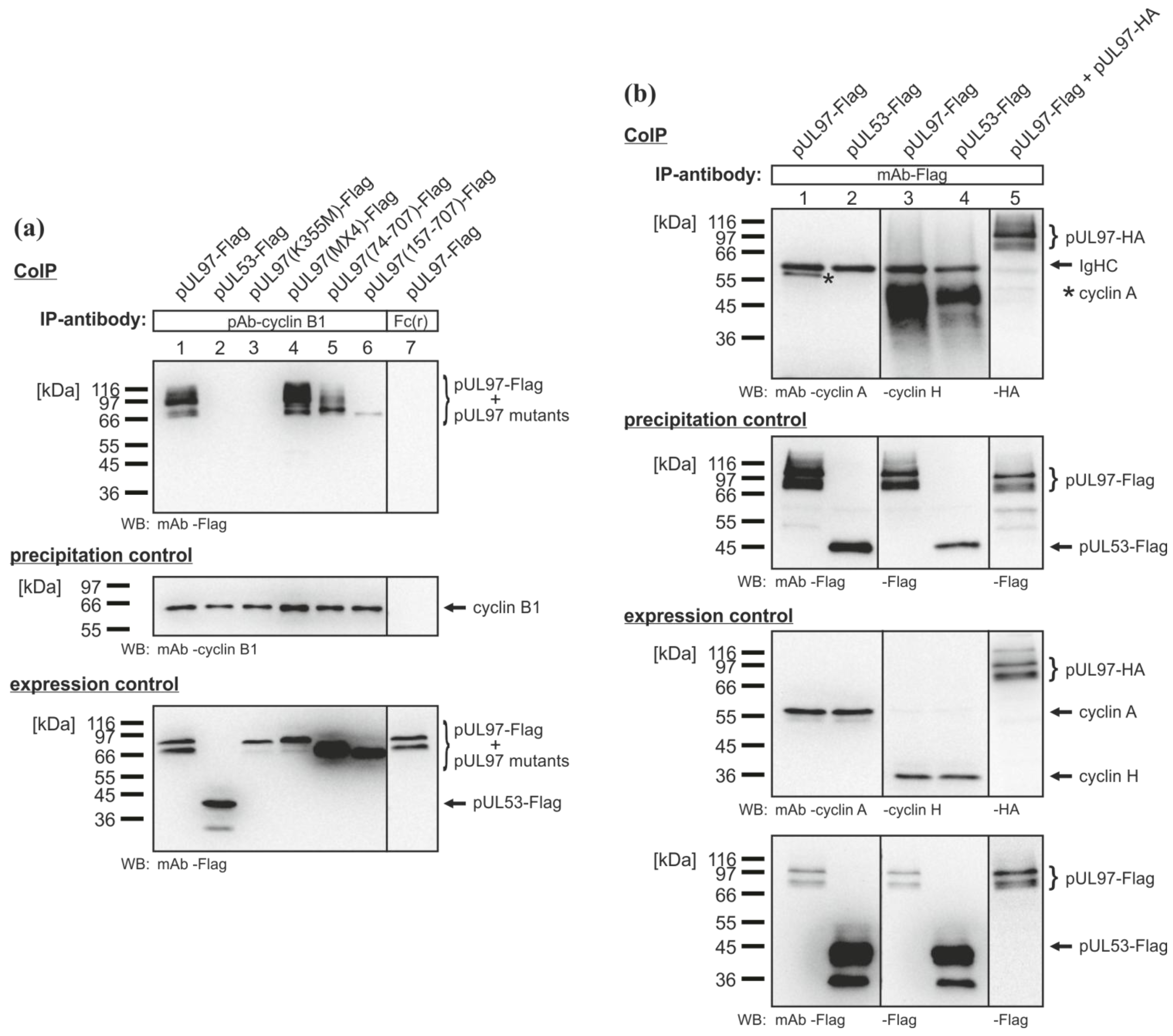
2.3. Mapping of the Sequence Domain of pUL97 Responsible for the Interaction with Cyclin T1
2.4. Interaction of the HCMV Protein Kinase pUL97 with Further Cyclins

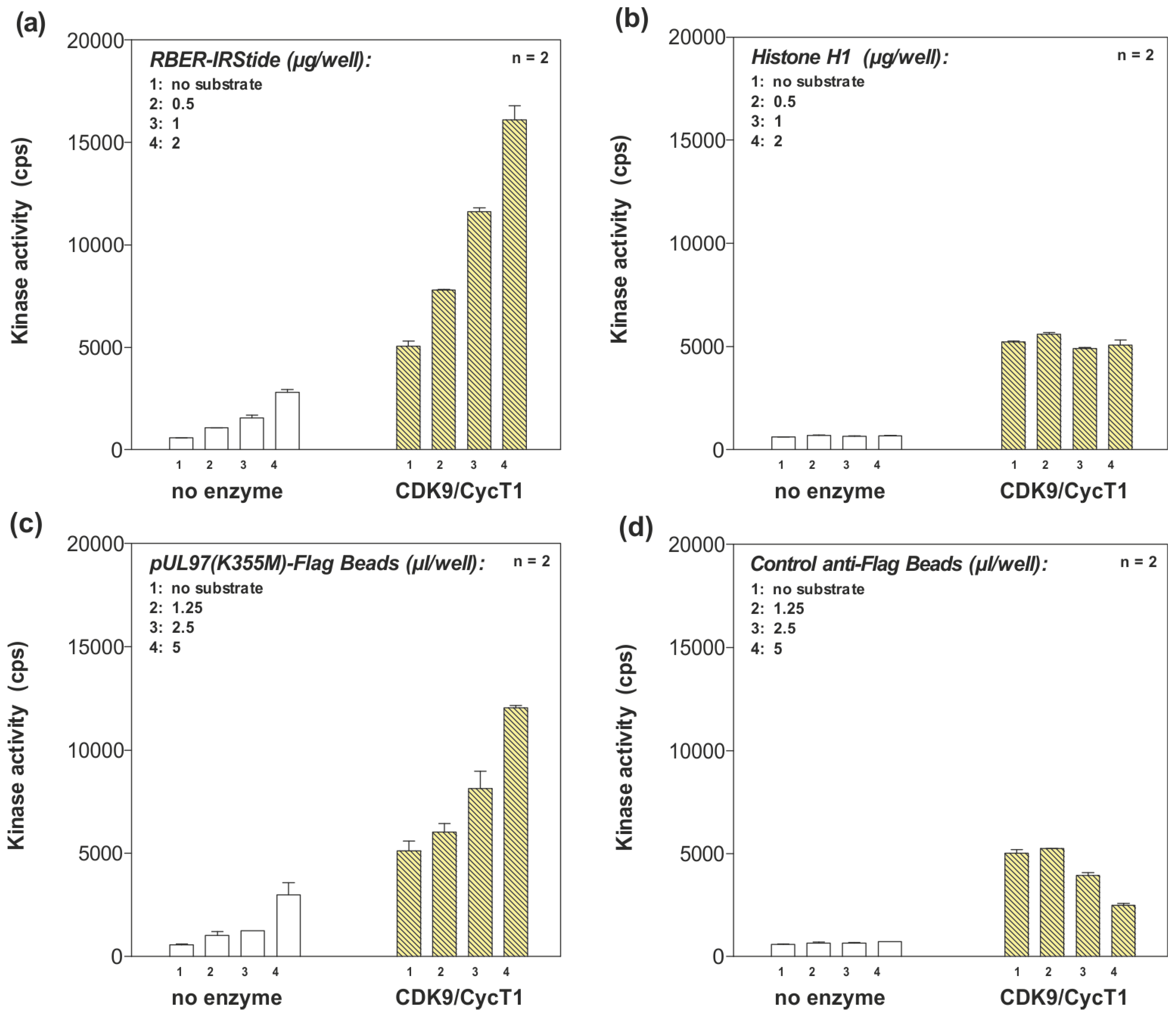
2.5. Phosphorylation of pUL97 by the CDK9/cyclin T1 Complex in an ATP Consumption Assay
3. Experimental Section
3.1. Cell Culture, HCMV Infections and Plasmid Transfection
3.2. Antibodies


3.3. Co-Immunoprecipitation
3.4. Immunofluorescence Analyses
3.5. ATP Consumption Assay
3.6. Yeast-Two-Hybrid System
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2701–2772. [Google Scholar]
- Schreiber, A.; Härter, G.; Schubert, A.; Bunjes, D.; Mertens, T.; Michel, D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin. Pharmacother. 2009, 10, 191–209. [Google Scholar] [CrossRef]
- Marschall, M.; Feichtinger, S.; Milbradt, J. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 2011, 80, 69–101. [Google Scholar] [CrossRef]
- Marschall, M.; Stamminger, T. Molecular targets for antiviral therapy of cytomegalovirus infections. Future Microbiol. 2009, 4, 731–742. [Google Scholar] [CrossRef]
- Prichard, M.N. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 2009, 19, 215–229. [Google Scholar] [CrossRef]
- Chou, S. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 2008, 18, 233–246. [Google Scholar] [CrossRef]
- Herget, T.; Marschall, M. Recent developments in anti-herpesviral combination therapy based on protein kinase inhibitors. In New Concepts of Antiviral Therapy; Bogner, E., Holzenburg, A., Eds.; Springer: London, UK, 2006; pp. 351–371. [Google Scholar]
- Schang, L.M.; St. Vincent, M.R.; Lacasse, J.J. Five years of progress on cyclindependent kinases and other cellular proteins as potential targets for antiviral drugs. Antivir. Chem. Chemother. 2006, 17, 293–320. [Google Scholar]
- Feichtinger, S.; Stamminger, T.; Müller, R.; Graf, L.; Klebl, B.; Eickhoff, J.; Marschall, M. Recruitment of cyclin-dependent kinase 9 to nuclear compartments during cytomegalovirus late replication: Importance of an interaction between viral pUL69 and cyclin T1. J. Gen. Virol. 2011, 92, 1519–1531. [Google Scholar] [CrossRef]
- Schang, L.M. First demonstration of the effectiveness of inhibitors of cellular protein kinases in antiviral therapy. Expert Rev. Anti Infect. Ther. 2006, 4, 953–956. [Google Scholar] [CrossRef]
- Sanchez, V.; McElroy, A.K.; Yen, J.; Tamrakar, S.; Clark, C.L.; Schwartz, R.A.; Spector, D.H. Cyclin-dependent kinase activity is required at early times for accurate processing and accumulation of the human cytomegalovirus UL122–123 and UL37 immediate-early transcripts and at later times for virus production. J. Virol. 2004, 78, 11219–11232. [Google Scholar] [CrossRef]
- Hutterer, C.; Wandinger, S.K.; Wagner, S.; Müller, R.; Stamminger, T.; Zeitträger, I.; Godl, K.; Baumgartner, R.; Strobl, S.; Marschall, M. Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases Aurora A, ABL and AMPK. Antivir. Res. 2013, 99, 139–148. [Google Scholar] [CrossRef]
- Kapasi, A.J.; Spector, D.H. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J. Virol. 2008, 82, 394–407. [Google Scholar] [CrossRef]
- Sanchez, V.; Spector, D.H. Cyclin-dependent kinase activity is required for efficient expression and posttranslational modification of human cytomegalovirus proteins and for production of extracellular particles. J. Virol. 2006, 80, 5886–5896. [Google Scholar] [CrossRef]
- Tamrakar, S.; Kapasi, A.J.; Spector, D.H. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 2005, 79, 15477–15493. [Google Scholar]
- Marschall, M.; Stein-Gerlach, M.; Freitag, M.; Kupfer, R.; van den Bogaard, M.; Stamminger, T. Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle of antiviral therapy. J. Gen. Virol. 2002, 83, 1013–1023. [Google Scholar]
- Prichard, M.N.; Gao, N.; Jairath, S.; Mulamba, G.; Krosky, P.; Coen, D.M.; Parker, B.O.; Pari, G.S. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 1999, 73, 5663–5670. [Google Scholar]
- Romaker, D.; Schregel, V.; Maurer, K.; Auerochs, S.; Marzi, A.; Sticht, H.; Marschall, M. Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem. 2006, 49, 7044–7053. [Google Scholar] [CrossRef]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, 1001092:1–1001092:17. [Google Scholar]
- Hume, A.J.; Finkel, J.S.; Kamil, J.P.; Coen, D.M.; Culbertson, M.R.; Kalejta, R.F. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 2008, 320, 797–799. [Google Scholar] [CrossRef]
- Prichard, M.N.; Sztul, E.; Daily, S.L.; Perry, A.L.; Frederick, S.L.; Gill, R.B.; Hartline, C.B.; Streblow, D.N.; Varnum, S.M.; Smith, R.D.; Kern, E.R. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 2008, 82, 5054–5067. [Google Scholar] [CrossRef]
- Hertel, L.; Chou, S.; Mocarski, E.S. Viral and cell cycle-regulated kinases in cytomegalovirus-induced pseudomitosis and replication. PLoS Pathog. 2007, 3, 6:0014–6:0022. [Google Scholar]
- Marschall, M.; Stein-Gerlach, M.; Freitag, M.; Kupfer, R.; van den Bogaard, M.; Stamminger, T. Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J. Gen. Virol. 2001, 82, 1439–1450. [Google Scholar]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar]
- Webel, R.; Solbak, S.M.Ø.; Fossen, T.; Auerochs, S.; Sticht, H.; Chou, S.; Marschall, M. Specification of the HCMV pUL97 isoforms: Differences in subcellular localization and functionality. In Proceedings of the 37th Annual International Herpesvirus Workshop, Calgary, Canada, 4–9 August 2012. No. 3.22.
- Webel, R.; Milbradt, J.; Auerochs, S.; Schregel, V.; Held, C.; Nöbauer, K.; Razzazi-Fazeli, E.; Jardin, C.; Wittenberg, T.; Sticht, H.; Marschall, M. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 2011, 92, 638–649. [Google Scholar] [CrossRef]
- Webel, R.; Hakki, M.; Prichard, M.; Rawlinson, D.W.; Marschall, M.; Chou, S. Differential properties of three isoforms of cytomegalovirus protein kinase pUL97 affect viral replication and maribavir susceptibility. J. Virol. 2013. to be submitted for publication. [Google Scholar]
- Webel, R.; Solbak, S.M.Ø.; Held, C.; Milbradt, J.; Groß, A.; Eichler, J.; Wittenberg, T.; Jardin, C.; Sticht, H.; Fossen, T.; Marschall, M. The nuclear import of isoforms of the cytomegalovirus kinase pUL97 is mediated by differential activity of NLS1 and NLS2 both acting through classical importin-α binding. J. Gen. Virol. 2012, 93, 1756–1768. [Google Scholar] [CrossRef]
- Schregel, V.; Auerochs, S.; Jochmann, R.; Maurer, K.; Stamminger, T.; Marschall, M. Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Virol. 2007, 88, 395–404. [Google Scholar] [CrossRef]
- Baek, M.C.; Krosky, P.M.; Pearson, A.; Coen, D.M. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 2004, 324, 184–193. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Matsumura, T.; Roizman, B.; Hirai, K. Cellular elongation factor 1δ is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 1999, 73, 4456–4460. [Google Scholar]
- Thomas, M.; Rechter, S.; Milbradt, J.; Auerochs, S.; Müller, R.; Stamminger, T.; Marschall, M. The cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. J. Gen. Virol. 2009, 90, 567–578. [Google Scholar] [CrossRef]
- Marschall, M.; Freitag, M.; Suchy, P.; Romaker, D.; Kupfer, R.; Hanke, M.; Stamminger, T. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 2003, 311, 60–71. [Google Scholar] [CrossRef]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef]
- Marschall, M.; Institute for Clinical and Molecular Virology, University of Erlangen-Nuremberg, Erlangen, Germany. p32/gC1qR, a multi-ligand binding protein. Unpublished work. 2013. [Google Scholar]
- Becke, S.; Fabre-Mersseman, V.; Aue, S.; Auerochs, S.; Sedmak, T.; Wolfrum, U.; Strand, D.; Marschall, M.; Plachter, B.; Reyda, S. Modification of the major tegument protein pp 65 of 1 human cytomegalovirus inhibits viral growth and leads to the enhancement of a protein complex with pUL69 and pUL97 in infected cells. J. Gen. Virol. 2010, 91, 2531–2541. [Google Scholar] [CrossRef]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar]
- Dinkel, H.; Michael, S.; Weatheritt, R.J.; Davey, N.E.; van Roey, K.; Altenberg, B.; Toedt, G.; Uyar, B.; Seiler, M.; Budd, A.; et al. ELM—The database of eukaryotic linear motifs. Nucleic Acids Res. 2012, 40 (Database issue), D242–D251. [Google Scholar]
- Marschall, M.; Freitag, M.; Weiler, S.; Sorg, G.; Stamminger, T. Recombinant green fluorescent protein-expressing human cytomegalovirus as a tool for screening antiviral agents. Antimicrob. Agents Chemother. 2000, 44, 1588–1597. [Google Scholar] [CrossRef]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Invest. 2010, 120, 3191–3208. [Google Scholar] [CrossRef]
- Winkler, M.; Rice, S.A.; Stamminger, T. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 1994, 68, 3943–3954. [Google Scholar]
- Milbradt, J.; Auerochs, S.; Marschall, M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007, 88, 2642–2650. [Google Scholar] [CrossRef]
- Fraldi, A.; Licciardo, P.; Majello, B.; Giordano, A.; Lania, L. Distinct regions of cyclinT1 are required for binding to CDK9 and for recruitment to the HIV-1 Tat/TAR complex. J. Cell. Biochem. Suppl. 2001, 36, 247–253. [Google Scholar]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [Google Scholar] [CrossRef]
- Marschall, M.; Sticht, H.; Institute for Clinical and Molecular Virology and Institute of Biochemistry, University of Erlangen-Nuremberg, Erlangen, Germany. Identification of motifs in pUL97 mediating cyclin interaction. Unpublished work. 2013. [Google Scholar]
- Rechter, S.; Scott, G.M.; Eickhoff, J.; Zielke, K.; Auerochs, S.; Müller, R.; Stamminger, T.; Rawlinson, W.D.; Marschall, M. Cyclin-dependent kinases phosphorylate the cytomegalovirus RNA export protein pUL69 to modulate its nuclear localization and activity. J. Biol. Chem. 2009, 284, 8605–8613. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Graf, L.; Webel, R.; Wagner, S.; Hamilton, S.T.; Rawlinson, W.D.; Sticht, H.; Marschall, M. The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins. Viruses 2013, 5, 3213-3230. https://doi.org/10.3390/v5123213
Graf L, Webel R, Wagner S, Hamilton ST, Rawlinson WD, Sticht H, Marschall M. The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins. Viruses. 2013; 5(12):3213-3230. https://doi.org/10.3390/v5123213
Chicago/Turabian StyleGraf, Laura, Rike Webel, Sabrina Wagner, Stuart T. Hamilton, William D. Rawlinson, Heinrich Sticht, and Manfred Marschall. 2013. "The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins" Viruses 5, no. 12: 3213-3230. https://doi.org/10.3390/v5123213
APA StyleGraf, L., Webel, R., Wagner, S., Hamilton, S. T., Rawlinson, W. D., Sticht, H., & Marschall, M. (2013). The Cyclin-Dependent Kinase Ortholog pUL97 of Human Cytomegalovirus Interacts with Cyclins. Viruses, 5(12), 3213-3230. https://doi.org/10.3390/v5123213