Intrathecal Humoral Immunity to Encephalitic RNA Viruses

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
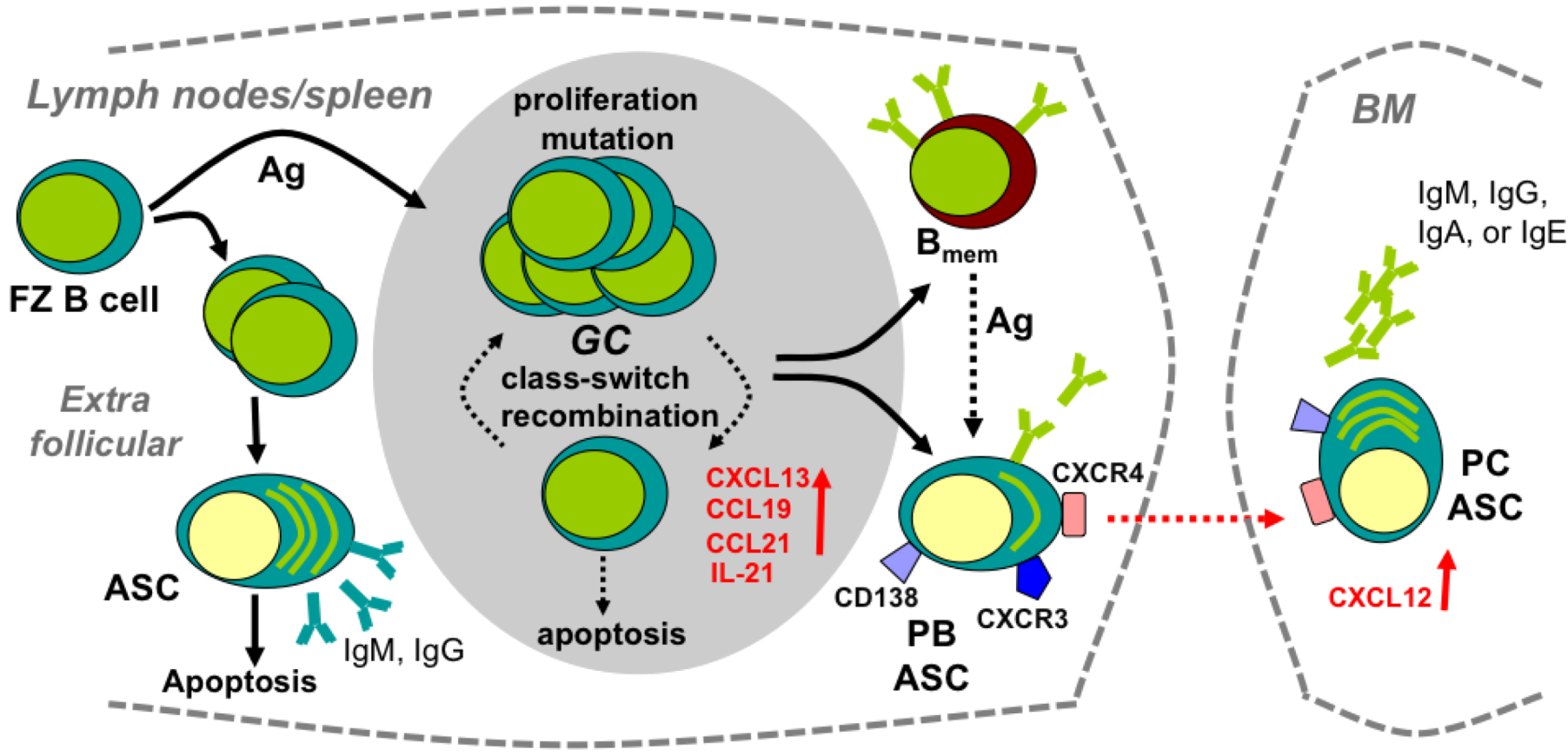
2. Development and Maintenance of B Cell Memory


3. Neurotropic Virus Models
4. Protective Mechanisms of Humoral Reponses in the CNS
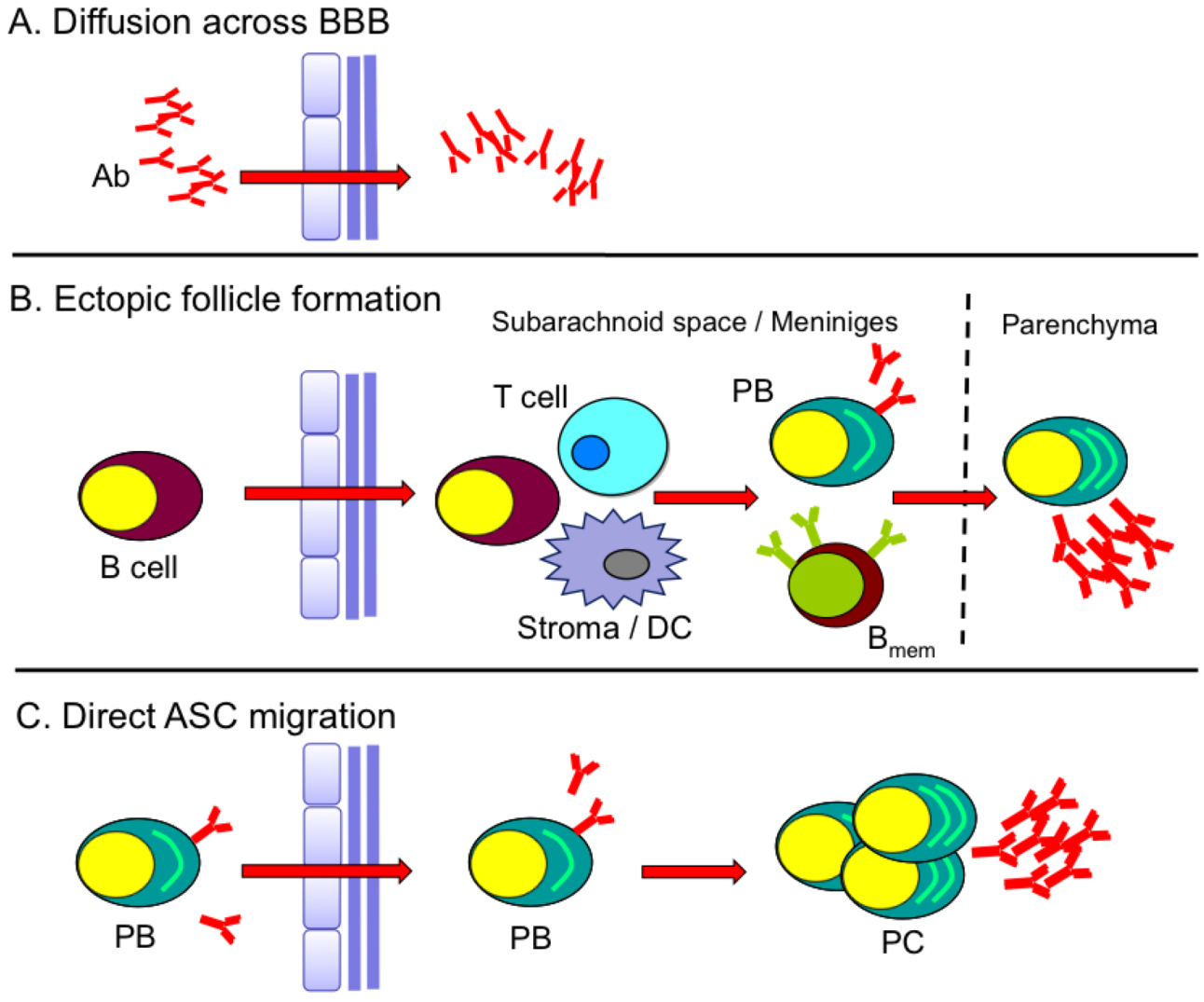
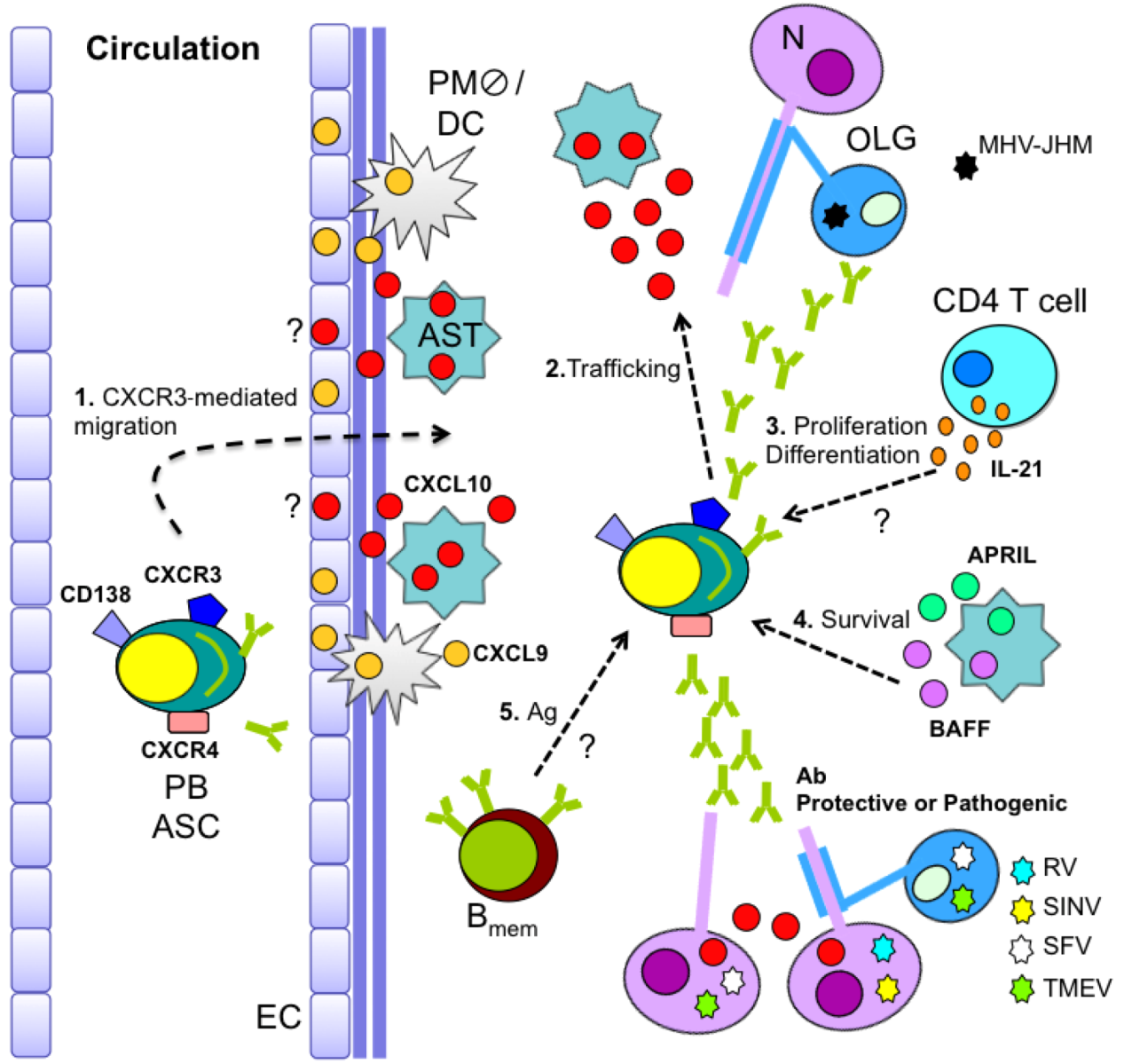
5. Kinetics of B Cell CNS Recruitment and Migration Signals
 = perivasacular macrophage; N = neuron; OLG = oligoendroglia
= perivasacular macrophage; N = neuron; OLG = oligoendroglia
= perivasacular macrophage; N = neuron; OLG = oligoendroglia
= perivasacular macrophage; N = neuron; OLG = oligoendroglia
6. ASC Differentiation and Survival in the CNS
7. Summary and Future Perspectives
Conflict of Interest
Acknowledgements
References
- McGavern, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol 2011, 11, 318–329. [Google Scholar] [CrossRef]
- Owens, G.P.; Gilden, D.; Burgoon, M.P.; Yu, X.; Bennett, J.L. Viruses and multiple sclerosis. Neurosci. 2011, 17, 659–676. [Google Scholar]
- Weissert, R. Progressive multifocal leukoencephalopathy. J. Neuroimmunol. 2011, 231, 73–77. [Google Scholar] [CrossRef]
- Wong, K.T. Emerging epidemic viral encephalitides with a special focus on henipaviruses. Acta Neuropath. 2010, 120, 317–325. [Google Scholar] [CrossRef]
- Johnson, N.; Cunningham, A.F.; Fooks, A.R. The immune response to rabies virus infection and vaccination. Vaccine 2010, 28, 3896–3901. [Google Scholar] [CrossRef]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Denizot, M.; Neal, J.W.; Gasque, P. Encephalitis due to emerging viruses: CNS innate immunity and potential therapeutic targets. J. Infect. 2012, 65, 1–16. [Google Scholar] [CrossRef]
- Skoldenberg, B.; Kalimo, K.; Carlstrom, A.; Forsgren, M.; Halonen, P. Herpes simplex encephalitis: A serological follow-up study. Synthesis of herpes simplex virus immunoglobulin M, A, and G antibodies and development of oligoclonal immunoglobulin G in the central nervous system. Acta Neurol. Scand. 1981, 63, 273–285. [Google Scholar]
- Schultze, D.; Weder, B.; Cassinotti, P.; Vitek, L.; Krausse, K.; Fierz, W. Diagnostic significance of intrathecally produced herpes simplex and varizella-zoster virus-specific antibodies in central nervous system infections. Swiss Med. 2004, 134, 700–704. [Google Scholar]
- Narayan, K.; Dail, D.; Li, L.; Cadavid, D.; Amrute, S.; Fitzgerald-Bocarsly, P.; Pachner, A.R. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann. Neurol. 2005, 57, 813–823. [Google Scholar] [CrossRef]
- Burke, D.S.; Nisalak, A.; Lorsomrudee, W.; Ussery, M.A.; Laorpongse, T. Virus-specific antibody-producing cells in blood and cerebrospinal fluid in acute Japanese encephalitis. J. Med. Virol. 1985, 17, 283–292. [Google Scholar] [CrossRef]
- Studahl, M.; Lindquist, L.; Eriksson, B.M.; Gunther, G.; Bengner, M.; Franzen-Rohl, E.; Fohlman, J.; Bergstrom, T.; Aurelius, E. Acute Viral Infections of the Central Nervous System in Immunocompetent Adults: Diagnosis and Management. Drugs 2013, (in press). [Google Scholar]
- Jacobi, C.; Lange, P.; Reiber, H. Quantitation of intrathecal antibodies in cerebrospinal fluid of subacute sclerosing panencephalitis, herpes simplex encephalitis and multiple sclerosis: discrimination between microorganism-driven and polyspecific immune response. J. Neuroimmunol. 2007, 187, 139–146. [Google Scholar] [CrossRef]
- Burke, D.S.; Nisalak, A.; Ussery, M.A.; Laorakpongse, T.; Chantavibul, S. Kinetics of IgM and IgG responses to Japanese encephalitis virus in human serum and cerebrospinal fluid. J. Infect. Dis. 1985, 151, 1093–1099. [Google Scholar] [CrossRef]
- Roivainen, M.; Agboatwalla, M.; Stenvik, M.; Rysa, T.; Akram, D.S.; Hovi, T. Intrathecal immune response and virus-specific immunoglobulin M antibodies in laboratory diagnosis of acute poliomyelitis. J. Clin. Microbiol. 1993, 31, 2427–2432. [Google Scholar]
- Kaiser, R.; Dorries, R.; Luer, W.; Poser, S.; Pohle, H.D.; Felgenhauer, K.; ter Meulen, V. Analysis of oligoclonal antibody bands against individual HIV structural proteins in the CSF of patients infected with HIV. J. Neurol. 1989, 236, 157–160. [Google Scholar] [CrossRef]
- Vandvik, B.; Weil, M.L.; Grandien, M.; Norrby, E. Progressive rubella virus panencephalitis: synthesis of oligoclonal virus-specific IgG antibodies and homogeneous free light chains in the central nervous system. Acta Neurol. Scand. 1978, 57, 53–64. [Google Scholar]
- Vandvik, B.; Norrby, E. Oligoclonal IgG antibody response in the central nervous system to different measles virus antigens in subacute sclerosing panencephalitis. Proc. Natl. Acad. Sci. USA 1973, 70, 1060–1063. [Google Scholar] [CrossRef]
- Thakare, J.P.; Gore, M.M.; Risbud, A.R.; Banerjee, K.; Ghosh, S.N. Detection of virus specific IgG subclasses in Japanese encephalitis patients. Indian J. Med. Res. 1991, 93, 271–276. [Google Scholar]
- Ryzhova, E.; Aye, P.; Harvey, T.; Cao, W.; Lackner, A.; Gonzalez-Scarano, F. Intrathecal humoral responses are inversely associated with the frequency of simian immunodeficiency virus macrophage-tropic variants in the central nervous system. J. Virol. 2009, 83, 8282–8288. [Google Scholar] [CrossRef]
- Puccioni-Sohler, M.; Rios, M.; Bianco, C.; Zhu, S.W.; Oliveira, C.; Novis, S.A.; Pombo-de-Oliveira, M.S. An inverse correlation of HTLV-I viral load in CSF and intrathecal synthesis of HTLV-I antibodies in TSP/HAM. Neurology 1999, 53, 1335–1339. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.; Costello, C.; Rosen-Schmidt, S.; Andersson, M.; Parks, D.; Perry, A.; Yerra, R.; Schmidt, R.; Alvarez, E.; Tyler, K.L. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch. Neurol. 2011, 68, 1156–1164. [Google Scholar] [CrossRef]
- Tavazzi, E.; Ferrante, P.; Khalili, K. Progressive multifocal leukoencephalopathy: An unexpected complication of modern therapeutic monoclonal antibody therapies. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases 2011, 17, 1776–1780. [Google Scholar]
- Griffin, D.; Levine, B.; Tyor, W.; Ubol, S.; Despres, P. The role of antibody in recovery from alphavirus encephalitis. Immunol. Rev. 1997, 159, 155–161. [Google Scholar] [CrossRef]
- Ramakrishna, C.; Bergmann, C.C.; Atkinson, R.; Stohlman, S.A. Control of central nervous system viral persistence by neutralizing antibody. J. Virol 2003, 77, 4670–4678. [Google Scholar] [CrossRef]
- Hooper, D.C.; Phares, T.W.; Fabis, M.J.; Roy, A. The production of antibody by invading B cells is required for the clearance of rabies virus from the central nervous system. PLoS Negl. Trop. Dis. 2009, 3, e535. [Google Scholar] [CrossRef]
- Marques, C.P.; Kapil, P.; Hinton, D.R.; Hindinger, C.; Nutt, S.L.; Ransohoff, R.M.; Phares, T.W.; Stohlman, S.A.; Bergmann, C.C. CXCR3-dependent plasma blast migration to the central nervous system during viral encephalomyelitis. J. Virol. 2011, 85, 6136–6147. [Google Scholar]
- Fragkoudis, R.; Ballany, C.M.; Boyd, A.; Fazakerley, J.K. In Semliki Forest virus encephalitis, antibody rapidly clears infectious virus and is required to eliminate viral material from the brain, but is not required to generate lesions of demyelination. J. Gen. Virol. 2008, 89, 2565–2568. [Google Scholar] [CrossRef]
- Levine, B.; Hardwick, J.M.; Trapp, B.D.; Crawford, T.O.; Bollinger, R.C.; Griffin, D.E. Antibody-mediated clearance of alphavirus infection from neurons. Science 1991, 254, 856–860. [Google Scholar]
- Pachner, A.R.; Brady, J.; Narayan, K. Antibody-secreting cells in the central nervous system in an animal model of MS: Phenotype, association with disability, and in vitro production of antibody. J. Neuroimmunol. 2007, 190, 112–120. [Google Scholar] [CrossRef]
- Lee, H.; Sunden, Y.; Ochiai, K.; Umemura, T. Experimental intracerebral vaccination protects mouse from a neurotropic virus by attracting antibody secreting cells to the CNS. Immunol. Lett. 2011, 139, 102–109. [Google Scholar] [CrossRef]
- Manz, R.A.; Hauser, A.E.; Hiepe, F.; Radbruch, A. Maintenance of serum antibody levels. Annu. Rev. Immunol. 2005, 23, 367–386. [Google Scholar] [CrossRef]
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol 2005, 5, 230–242. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Zotos, D.; Tarlinton, D.M. Determining germinal centre B cell fate. Trends Immunol. 2012, 33, 281–288. [Google Scholar] [CrossRef]
- Crotty, S. The 1–1-1 fallacy. Immunol. Rev. 2012, 247, 133–42. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Weisel, F. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef]
- Cyster, J.G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [Google Scholar] [CrossRef]
- Okada, T.; Cyster, J.G. B cell migration and interactions in the early phase of antibody responses. Curr. Opin. Immunol. 2006, 18, 278–285. [Google Scholar] [CrossRef]
- Muehlinghaus, G.; Cigliano, L.; Huehn, S.; Peddinghaus, A.; Leyendeckers, H.; Hauser, A.E.; Hiepe, F.; Radbruch, A.; Arce, S.; Manz, R.A. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood 2005, 105, 3965–3971. [Google Scholar]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dorner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef]
- Dorner, T.; Radbruch, A. Antibodies and B cell memory in viral immunity. Immunity 2007, 27, 384–392. [Google Scholar] [CrossRef]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar]
- Elgueta, R.; de Vries, V.C.; Noelle, R.J. The immortality of humoral immunity. Immunol. Rev 2010, 236, 139–150. [Google Scholar] [CrossRef]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of humoral immunity to common viral and vaccine antigens. New. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Megret, J.; Storck, S.; Reynaud, C.A.; Weill, J.C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Corcione, A.; Casazza, S.; Ferretti, E.; Giunti, D.; Zappia, E.; Pistorio, A.; Gambini, C.; Mancardi, G.L.; Uccelli, A.; Pistoia, V. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 11064–11069. [Google Scholar]
- Owens, G.P.; Bennett, J.L.; Gilden, D.H.; Burgoon, M.P. The B cell response in multiple sclerosis. Neurol. Res. 2006, 28, 236–244. [Google Scholar] [CrossRef]
- Owens, G.P.; Ritchie, A.M.; Burgoon, M.P.; Williamson, R.A.; Corboy, J.R.; Gilden, D.H. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J. Immunol 2003, 171, 2725–2733. [Google Scholar]
- Krumbholz, M.; Theil, D.; Derfuss, T.; Rosenwald, A.; Schrader, F.; Monoranu, C.M.; Kalled, S.L.; Hess, D.M.; Serafini, B.; Aloisi, F.; Wekerle, H.; Hohlfeld, R.; Meinl, E. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med. 2005, 201, 195–200. [Google Scholar] [CrossRef]
- Thangarajh, M.; Masterman, T.; Hillert, J.; Moerk, S.; Jonsson, R. A proliferation-inducing ligand (APRIL) is expressed by astrocytes and is increased in multiple sclerosis. Scand. J. Immunol. 2007, 65, 92–98. [Google Scholar]
- Phares, T.W.; Marques, C.P.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J. Virol. 2011, 85, 2589–2598. [Google Scholar] [CrossRef]
- Reiber, H.; Peter, J.B. Cerebrospinal fluid analysis: Disease-related data patterns and evaluation programs. J. Neurol. Sci. 2001, 184, 101–122. [Google Scholar] [CrossRef]
- Griffin, D.E. Recovery from viral encephalomyelitis: immune-mediated noncytolytic virus clearance from neurons. Immunol. Res. 2010, 47, 123–133. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus infection of the central nervous system: host-virus stand-off. Nature Reviews. Microbiol. 2006, 4, 121–132. [Google Scholar] [CrossRef]
- Lane, T.E.; Hosking, M.P. The pathogenesis of murine coronavirus infection of the central nervous system. Crit. Rev. Immunol. 2010, 30, 119–130. [Google Scholar] [CrossRef]
- Bender, S.J.; Weiss, S.R. Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune. Pharmacol. 2010, 5, 336–354. [Google Scholar] [CrossRef]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Atkinson, R.; Bergmann, C.C. Enhanced antiviral T cell function in the absence of B7-H1 is insufficient to prevent persistence but exacerbates axonal bystander damage during viral encephalomyelitis. J. Immunol. 2010, 185, 5607–5618. [Google Scholar] [CrossRef]
- Phares, T.W.; Stohlman, S.A.; Hwang, M.; Min, B.; Hinton, D.R.; Bergmann, C.C. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J. Virol. 2012, 86, 2416–2427. [Google Scholar] [CrossRef]
- Ramakrishna, C.; Stohlman, S.A.; Atkinson, R.D.; Shlomchik, M.J.; Bergmann, C.C. Mechanisms of central nervous system viral persistence: the critical role of antibody and B cells. J. Immunol. 2002, 168, 1204–1211. [Google Scholar]
- Griffin, D.E.; Metcalf, T. Clearance of virus infection from the CNS. Curr. Opin. Virol. 2011, 1, 216–221. [Google Scholar]
- Kurkela, S.; Manni, T.; Vaheri, A.; Vapalahti, O. Causative agent of Pogosta disease isolated from blood and skin lesions. Emerg. Infect. Dis. 2004, 10, 889–894. [Google Scholar] [CrossRef]
- Malherbe, H.; Strickland-Cholmley, M.; Jackson, A.L. Sindbis virus infection in man. Report of a case with recovery of virus from skin lesions. S. Afr. Med. J. 1963, 37, 547–552. [Google Scholar]
- Luukkainen, R.; Laine, M.; Nirhamo, J. Chronic arthritis after Sindbis-related (Pogosta) virus infection. Scand. J. Rheumatol. 2000, 29, 399–400. [Google Scholar] [CrossRef]
- Jackson, A.C.; Moench, T.R.; Griffin, D.E.; Johnson, R.T. The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab. Invest. 1987, 56, 418–423. [Google Scholar]
- Griffin, D.E. Role of the immune response in age-dependent resistance of mice to encephalitis due to Sindbis virus. J. Infect. Dis. 1976, 133, 456–464. [Google Scholar] [CrossRef]
- Havert, M.B.; Schofield, B.; Griffin, D.E.; Irani, D.N. Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted sindbis virus infection of mice. J. Virol. 2000, 74, 5352–5356. [Google Scholar] [CrossRef]
- Lewis, J.; Wesselingh, S.L.; Griffin, D.E.; Hardwick, J.M. Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J. Virol. 1996, 70, 1828–1835. [Google Scholar]
- Burdeinick-Kerr, R.; Griffin, D.E. Gamma interferon-dependent, noncytolytic clearance of sindbis virus infection from neurons in vitro. J. Virol. 2005, 79, 5374–5385. [Google Scholar] [CrossRef]
- Nava, V.E.; Rosen, A.; Veliuona, M.A.; Clem, R.J.; Levine, B.; Hardwick, J.M. Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J. Virol. 1998, 72, 452–459. [Google Scholar]
- Metcalf, T.U.; Griffin, D.E. Alphavirus-induced encephalomyelitis: Antibody-secreting cells and viral clearance from the nervous system. J. Virol. 2011, 85, 11490–11501. [Google Scholar] [CrossRef]
- Balluz, I.M.; Glasgow, G.M.; Killen, H.M.; Mabruk, M.J.; Sheahan, B.J.; Atkins, G.J. Virulent and avirulent strains of Semliki Forest virus show similar cell tropism for the murine central nervous system but differ in the severity and rate of induction of cytolytic damage. Neuropathol. Appl. Neurobiol. 1993, 19, 233–239. [Google Scholar] [CrossRef]
- Fazakerley, J.K. Pathogenesis of Semliki Forest virus encephalitis. J. Neurovirol. 2002, 2, 66–74. [Google Scholar] [CrossRef]
- Mathiot, C.C.; Grimaud, G.; Garry, P.; Bouquety, J.C.; Mada, A.; Daguisy, A.M.; Georges, A.J. An outbreak of human Semliki Forest virus infections in Central African Republic. Am. J. Trop. Med. Hyg. 1990, 42, 386–3893. [Google Scholar]
- Willems, W.R.; Kaluza, G.; Boschek, C.B.; Bauer, H.; Hager, H.; Schutz, H.J.; Feistner, H. Semliki forest virus: Cause of a fatal case of human encephalitis. Science 1979, 203, 1127–1129. [Google Scholar]
- Warrell, M.J. Current rabies vaccines and prophylaxis schedules: preventing rabies before and after exposure. Travel. Med. Infect. Dis. 2012, 10, 1–15. [Google Scholar] [CrossRef]
- Hooper, D.C.; Morimoto, K.; Bette, M.; Weihe, E.; Koprowski, H.; Dietzschold, B. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J. Virol. 1998, 72, 3711–3719. [Google Scholar]
- Tsunoda, I.; Fujinami, R.S. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler's murine encephalomyelitis virus. J. Neuropathol. Exp. Neurol. 1996, 55, 673–686. [Google Scholar] [CrossRef]
- Aubert, C.; Brahic, M. Early infection of the central nervous system by the GDVII and DA strains of Theiler's virus. J. Virol. 1995, 69, 3197–3200. [Google Scholar]
- Simas, J.P.; Dyson, H.; Fazakerley, J.K. The neurovirulent GDVII strain of Theiler's virus can replicate in glial cells. J. Virol. 1995, 69, 5599–5606. [Google Scholar]
- Lipton, H.L.; Kumar, A.S.; Trottier, M. Theiler's virus persistence in the central nervous system of mice is associated with continuous viral replication and a difference in outcome of infection of infiltrating macrophages versus oligodendrocytes. Virus. Res. 2005, 111, 214–223. [Google Scholar] [CrossRef]
- Matthews, A.E.; Weiss, S.R.; Shlomchik, M.J.; Hannum, L.G.; Gombold, J.L.; Paterson, Y. Antibody is required for clearance of infectious murine hepatitis virus A59 from the central nervous system, but not the liver. J. Immunol. 2001, 167, 5254–5263. [Google Scholar]
- Tyor, W.R.; Wesselingh, S.; Levine, B.; Griffin, D.E. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1992, 149, 4016–4020. [Google Scholar]
- Levine, B.; Griffin, D.E. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J. Virol. 1992, 66, 6429–6435. [Google Scholar]
- Hooper, D.C.; Roy, A.; Kean, R.B.; Phares, T.W.; Barkhouse, D.A. Therapeutic immune clearance of rabies virus from the CNS. Future Virol. 2011, 6, 387–397. [Google Scholar] [CrossRef]
- Phares, T.W.; Kean, R.B.; Mikheeva, T.; Hooper, D.C. Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J. Immunol. 2006, 176, 7666–7675. [Google Scholar]
- Fabis, M.J.; Phares, T.W.; Kean, R.B.; Koprowski, H.; Hooper, D.C. Blood-brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2008, 105, 15511–15516. [Google Scholar]
- Ubol, S.; Levine, B.; Lee, S.H.; Greenspan, N.S.; Griffin, D.E. Roles of immunoglobulin valency and the heavy-chain constant domain in antibody-mediated downregulation of Sindbis virus replication in persistently infected neurons. J. Virol. 1995, 69, 1990–1993. [Google Scholar]
- Despres, P.; Griffin, J.W.; Griffin, D.E. Antiviral activity of alpha interferon in Sindbis virus-infected cells is restored by anti-E2 monoclonal antibody treatment. J. Virol. 1995, 69, 7345–7348. [Google Scholar]
- Despres, P.; Griffin, J.W.; Griffin, D.E. Effects of anti-E2 monoclonal antibody on sindbis virus replication in AT3 cells expressing bcl-2. J. Virol. 1995, 69, 7006–7014. [Google Scholar]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef]
- Dietzschold, B.; Tollis, M.; Lafon, M.; Wunner, W.H.; Koprowski, H. Mechanisms of rabies virus neutralization by glycoprotein-specific monoclonal antibodies. Virology 1987, 161, 29–36. [Google Scholar] [CrossRef]
- Dietzschold, B. Antibody-mediated clearance of viruses from the mammalian central nervous system. T.I.M. 1993, 1((2)), 63–66. [Google Scholar]
- Dietzschold, B.; Kao, M.; Zheng, Y.M.; Chen, Z.Y.; Maul, G.; Fu, Z.F.; Rupprecht, C.E.; Koprowski, H. Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 7252–7256. [Google Scholar]
- Leibowitz, J.L.; Bond, C.W.; Anderson, K.; Goss, S. Biological and macromolecular properties of murine cells persistently infected with MHV-JHM. Arch. Virol. 1984, 80, 315–332. [Google Scholar] [CrossRef]
- Smith-Norowitz, T.A.; Sobel, R.A.; Mokhtarian, F. B cells and antibodies in the pathogenesis of myelin injury in Semliki Forest Virus encephalomyelitis. Cell. Immunol. 2000, 200, 27–35. [Google Scholar] [CrossRef]
- Subak-Sharpe, I.; Dyson, H.; Fazakerley, J. In vivo depletion of CD8+ T cells prevents lesions of demyelination in Semliki Forest virus infection. J. Virol. 1993, 67, 7629–7633. [Google Scholar]
- Mokhtarian, F.; Huan, C.M.; Roman, C.; Raine, C.S. Semliki Forest virus-induced demyelination and remyelination--involvement of B cells and anti-myelin antibodies. J. Neuroimmunol. 2003, 137, 19–31. [Google Scholar] [CrossRef]
- Knopf, P.M.; Harling-Berg, C.J.; Lee, D.J.; Hallett, J.J.; Stopa, E.G.; Mokhtarian, F. Microinfusion into the rat brain of antibodies against Semliki Forest Virus produces changes in behavioral response to apomorphine. J. Neuroimmunol. 2007, 184, 149–155. [Google Scholar] [CrossRef]
- Mokhtarian, F.; Safavi, F.; Sarafraz-Yazdi, E. Immunization with a peptide of Semliki Forest virus promotes remyelination in experimental autoimmune encephalomyelitis. Brain Res. 2012, 1488, 92–103. [Google Scholar] [CrossRef]
- Safavi, F.; Feliberti, J.P.; Raine, C.S.; Mokhtarian, F. Role of gammadelta T cells in antibody production and recovery from SFV demyelinating disease. J. Neuroimmunol. 2011, 235, 18–26. [Google Scholar] [CrossRef]
- Kang, B.S.; Palma, J.P.; Lyman, M.A.; Dal Canto, M.; Kim, B.S. Antibody response is required for protection from Theiler's virus-induced encephalitis in C57BL/6 mice in the absence of CD8+ T cells. Virology 2005, 340, 84–94. [Google Scholar] [CrossRef]
- Kurtz, C.I.; Sun, X.M.; Fujinami, R.S. B-lymphocyte requirement for vaccine-mediated protection from Theiler's murine encephalomyelitis virus-induced central nervous system disease. J. Virol. 1995, 69, 5152–5155. [Google Scholar]
- Cash, E.; Bandeira, A.; Chirinian, S.; Brahic, M. Characterization of B lymphocytes present in the demyelinating lesions induced by Theiler's virus. J. Immunol. 1989, 143, 984–988. [Google Scholar]
- Dal Canto, M.C.; Barbano, R.L. Immunocytochemical localization of MAG, MBP and P0 protein in acute and relapsing demyelinating lesions of Theiler's virus infection. J. Neuroimmunol. 1985, 10, 129–140. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Barbano, R.L. Antibody to myelin-associated glycoprotein reacts with plasma cells in mice. J. Neuroimmunol. 1986, 10, 279–286. [Google Scholar] [CrossRef]
- Tschen, S.I.; Bergmann, C.C.; Ramakrishna, C.; Morales, S.; Atkinson, R.; Stohlman, S.A. Recruitment kinetics and composition of antibody-secreting cells within the central nervous system following viral encephalomyelitis. J. Immunol. 2002, 168, 2922–2929. [Google Scholar]
- Tschen, S.I.; Stohlman, S.A.; Ramakrishna, C.; Hinton, D.R.; Atkinson, R.D.; Bergmann, C.C. CNS viral infection diverts homing of antibody-secreting cells from lymphoid organs to the CNS. Eur. J. Immunol. 2006, 36, 603–612. [Google Scholar] [CrossRef]
- Gil-Cruz, C.; Perez-Shibayama, C.; Firner, S.; Waisman, A.; Bechmann, I.; Thiel, V.; Cervantes-Barragan, L.; Ludewig, B. T helper cell- and CD40-dependent germline IgM prevents chronic virus-induced demyelinating disease. Proc. Natl. Acad. Sci. USA 2012, 109, 1233–1238. [Google Scholar]
- Griffin, D.E. Immunoglobulins in the cerebrospinal fluid: Changes during acute viral encephalitis in mice. J. Immunol. 1981, 126, 27–31. [Google Scholar]
- Parsons, L.M.; Webb, H.E. Virus titres and persistently raised white cell counts in cerebrospinal fluid in mice after peripheral infection with demyelinating Semliki Forest virus. Neuropathol. Appl. Neurobiol. 1982, 8, 395–401. [Google Scholar] [CrossRef]
- Parsons, L.M.; Webb, H.E. Blood brain barrier disturbance and immunoglobulin G levels in the cerebrospinal fluid of the mouse following peripheral infection with the demyelinating strain of Semliki Forest virus. J. Neurol. Sci. 1982, 57, 307–318. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Geuking, M.B.; McCoy, K.D. Homeland security: IgA immunity at the frontiers of the body. Trends Immunol. 2012, 33, 160–167. [Google Scholar]
- Oh, J.W.; Schwiebert, L.M.; Benveniste, E.N. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J. Neurovirol. 1999, 5, 82–94. [Google Scholar]
- Cole, K.E.; Strick, C.A.; Paradis, T.J.; Ogborne, K.T.; Loetscher, M.; Gladue, R.P.; Lin, W.; Boyd, J.G.; Moser, B.; Wood, D.E.; Sahagan, B.G.; Neote, K. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998, 187, 2009–2021. [Google Scholar]
- Luster, A.D.; Ravetch, J.V. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J. Exp. Med. 1987, 166, 1084–1097. [Google Scholar]
- Lane, T.E.; Asensio, V.C.; Yu, N.; Paoletti, A.D.; Campbell, I.L.; Buchmeier, M.J. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 1998, 160, 970–978. [Google Scholar]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Astrocyte dervived CXCL10 drives accumulation of antibody secreting cells in the centrtal nervous system during viral encephalomyelitis. J. Virol. 2013, in press. [Google Scholar]
- Hauser, A.E.; Debes, G.F.; Arce, S.; Cassese, G.; Hamann, A.; Radbruch, A.; Manz, R.A. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J. Immunol. 2002, 169, 1277–1282. [Google Scholar]
- Metcalf, T.U.; Baxter, V.K.; Nilaratanakul, V.; Griffin, D.E. Recruitment and Retention of B Cells in the CNS in Response to Alphavirus Encephalomyelitis. J. Virol. 2013, 87, 2420–2429. [Google Scholar]
- Rainey-Barger, E.K.; Rumble, J.M.; Lalor, S.J.; Esen, N.; Segal, B.M.; Irani, D.N. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav. Immun. 2011, 25, 922–931. [Google Scholar]
- Kuang, Y.; Lackay, S.N.; Zhao, L.; Fu, Z.F. Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after rabies virus infection. Virus Res. 2009, 144, 18–26. [Google Scholar]
- Nakamichi, K.; Saiki, M.; Sawada, M.; Takayama-Ito, M.; Yamamuro, Y.; Morimoto, K.; Kurane, I. Rabies virus-induced activation of mitogen-activated protein kinase and NF-kappaB signaling pathways regulates expression of CXC and CC chemokine ligands in microglia. J. Virol. 2005, 79, 11801–11812. [Google Scholar]
- Hoffman, L.M.; Fife, B.T.; Begolka, W.S.; Miller, S.D.; Karpus, W.J. Central nervous system chemokine expression during Theiler's virus-induced demyelinating disease. J. Neurovirol. 1999, 5, 635–642. [Google Scholar]
- Murray, P.D.; Krivacic, K.; Chernosky, A.; Wei, T.; Ransohoff, R.M.; Rodriguez, M. Biphasic and regionally-restricted chemokine expression in the central nervous system in the Theiler's virus model of multiple sclerosis. J. Neurovirol. 2000, 6, 44–52. [Google Scholar]
- Tsunoda, I.; Lane, T.E.; Blackett, J.; Fujinami, R.S. Distinct roles for IP-10/CXCL10 in three animal models, Theiler's virus infection, EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP-10. Mult. Scler. 2004, 10, 26–34. [Google Scholar]
- Ireland, D.D.; Stohlman, S.A.; Hinton, D.R.; Atkinson, R.; Bergmann, C.C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008, 82, 300–310. [Google Scholar]
- Palma, J.P.; Kim, B.S. Induction of selected chemokines in glial cells infected with Theiler's virus. J. Neuroimmunol. 2001, 117, 1, 166–170. [Google Scholar]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar]
- Magliozzi, R.; Columba-Cabezas, S.; Serafini, B.; Aloisi, F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004, 148, 11–23. [Google Scholar]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar]
- Serafini, B.; Severa, M.; Columba-Cabezas, S.; Rosicarelli, B.; Veroni, C.; Chiappetta, G.; Magliozzi, R.; Reynolds, R.; Coccia, E.M.; Aloisi, F. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J. Neuropathol. Exp. Neurol. 2010, 69, 677–693. [Google Scholar]
- Xu, W.; Joo, H.; Clayton, S.; Dullaers, M.; Herve, M.C.; Blankenship, D.; De La Morena, M.T.; Balderas, R.; Picard, C.; Casanova, J.L.; Pascual, V.; Oh, S.; Banchereau, J. Macrophages induce differentiation of plasma cells through CXCL10/IP-10. J. Exp. Med. 2012, 209, 1813–1823. [Google Scholar]
- Puntambekar, S.S.; Bergmann, C.C.; Savarin, C.; Karp, C.L.; Phares, T.W.; Parra, G.I.; Hinton, D.R.; Stohlman, S.A. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J. Virol. 2011, 85, 6702–6713. [Google Scholar]
- Linterman, M.A.; Beaton, L.; Yu, D.; Ramiscal, R.R.; Srivastava, M.; Hogan, J.J.; Verma, N.K.; Smyth, M.J.; Rigby, R.J.; Vinuesa, C.G. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010, 207, 353–363. [Google Scholar]
- Zotos, D.; Coquet, J.M.; Zhang, Y.; Light, A.; D'Costa, K.; Kallies, A.; Corcoran, L.M.; Godfrey, D.I.; Toellner, K.M.; Smyth, M.J.; Nutt, S.L.; Tarlinton, D.M. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 2010, 207, 365–378. [Google Scholar]
- Sabat, R.; Grutz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar]
- Aksamit, A.J. Review of progressive multifocal leukoencephalopathy and natalizumab. Neurologist 2006, 12, 293–298. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Phares, T.W.; Stohlman, S.A.; Bergmann, C.C. Intrathecal Humoral Immunity to Encephalitic RNA Viruses. Viruses 2013, 5, 732-752. https://doi.org/10.3390/v5020732
Phares TW, Stohlman SA, Bergmann CC. Intrathecal Humoral Immunity to Encephalitic RNA Viruses. Viruses. 2013; 5(2):732-752. https://doi.org/10.3390/v5020732
Chicago/Turabian StylePhares, Timothy W., Stephen A. Stohlman, and Cornelia C. Bergmann. 2013. "Intrathecal Humoral Immunity to Encephalitic RNA Viruses" Viruses 5, no. 2: 732-752. https://doi.org/10.3390/v5020732
APA StylePhares, T. W., Stohlman, S. A., & Bergmann, C. C. (2013). Intrathecal Humoral Immunity to Encephalitic RNA Viruses. Viruses, 5(2), 732-752. https://doi.org/10.3390/v5020732