Oxidative Stress and HPV Carcinogenesis

Abstract
:List of acronyms used

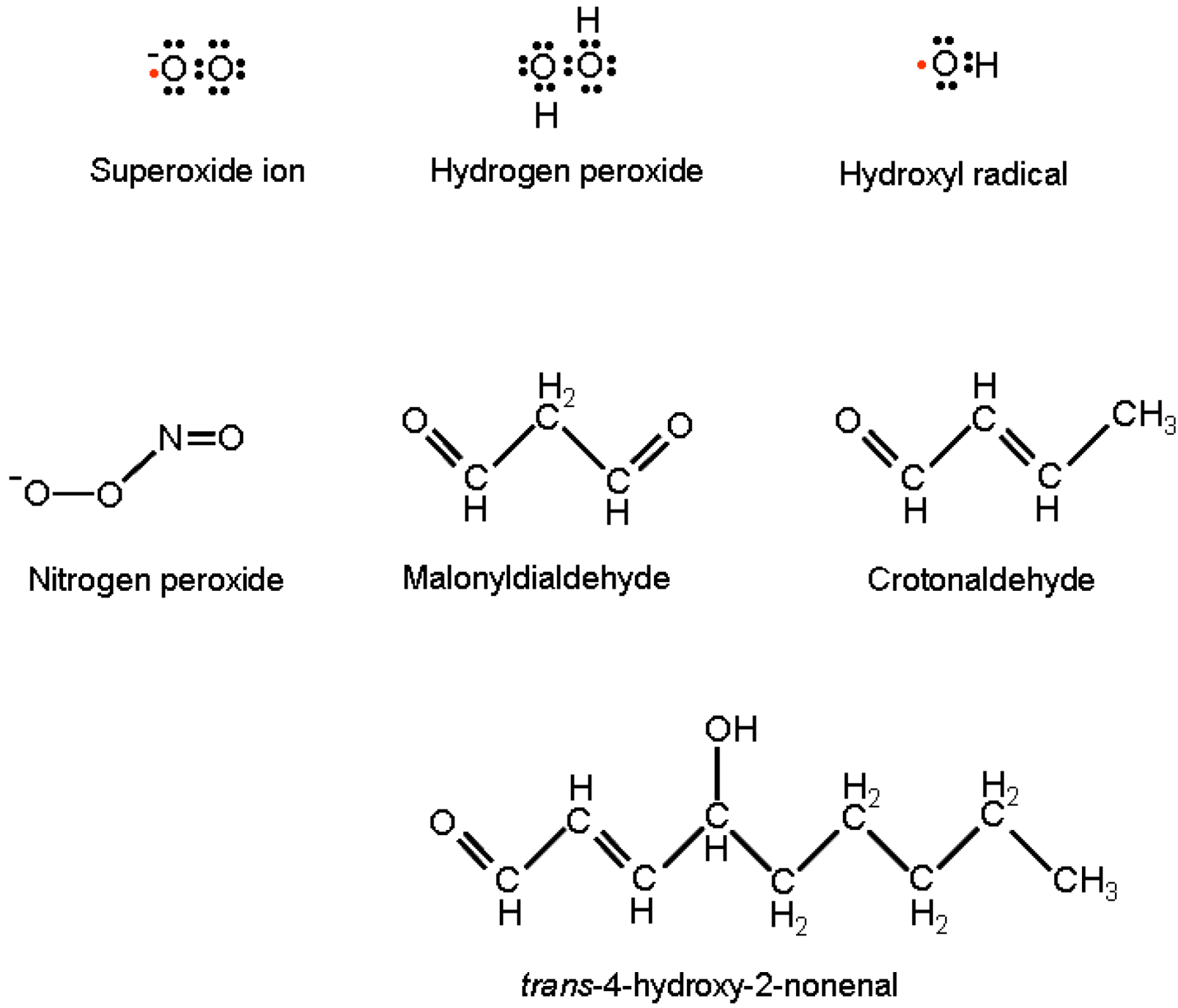{kind=link}
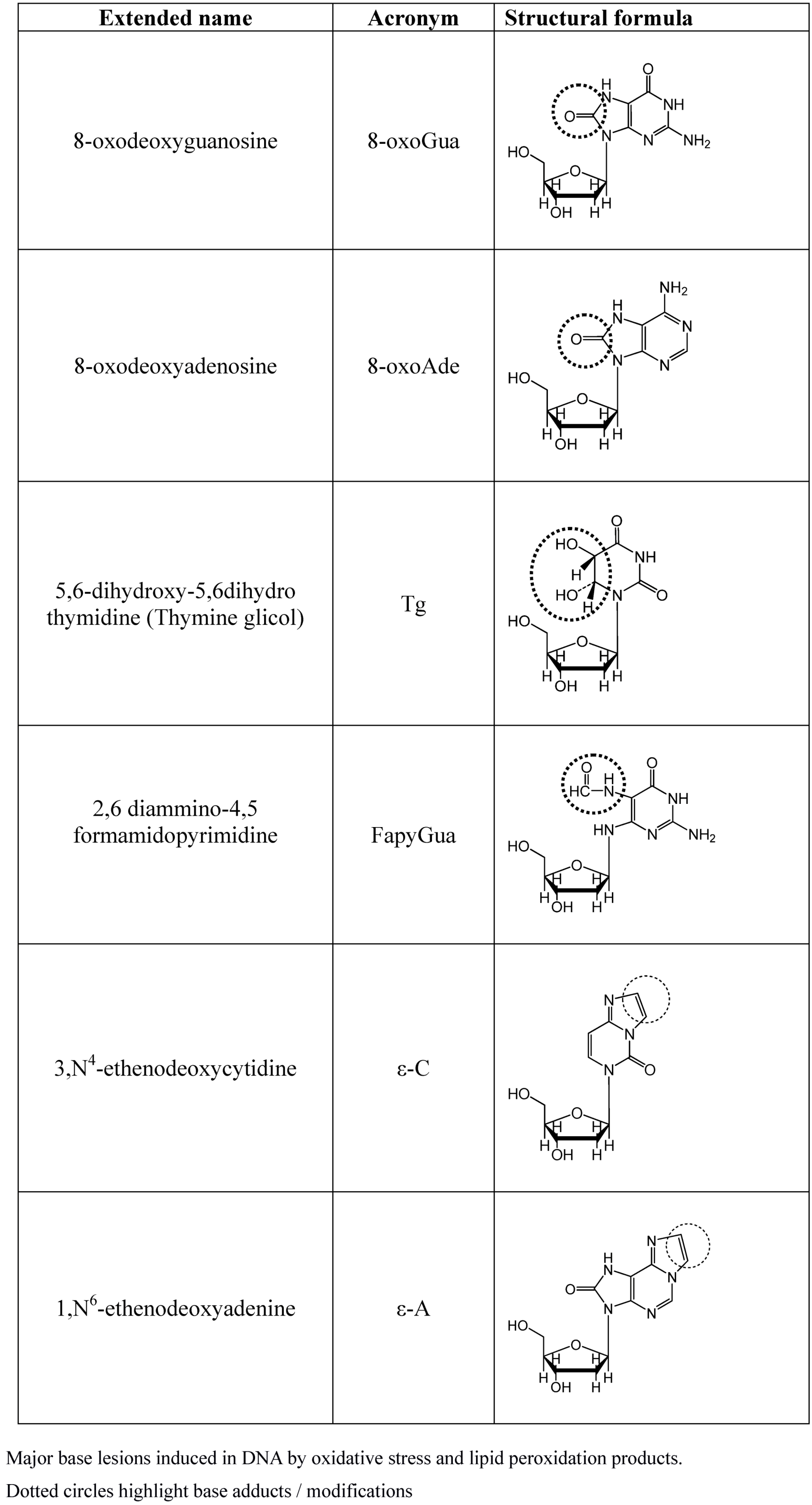{kind=link}
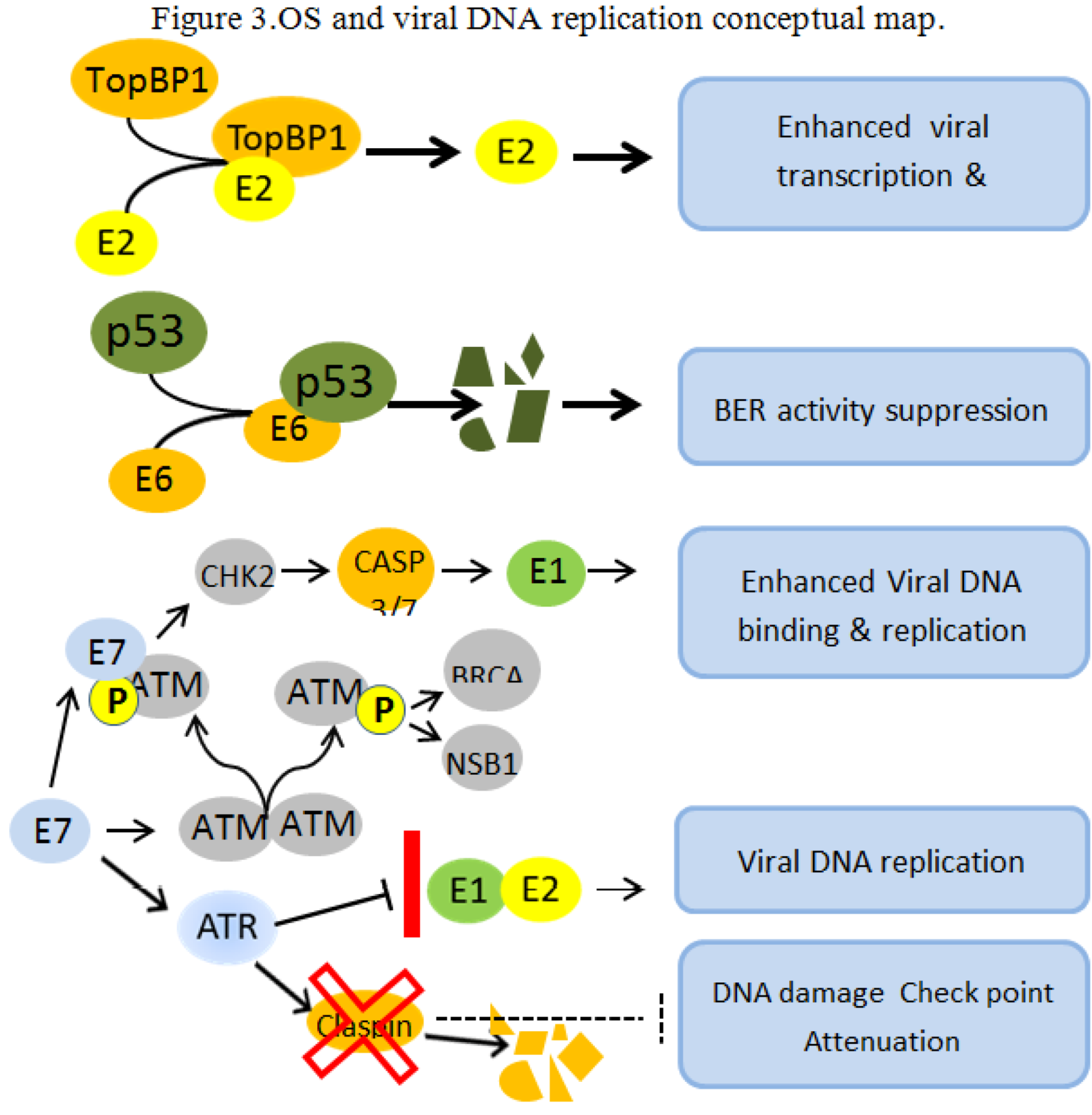{kind=link}
| AP-1 | Activator Protein 1 |
| ATM | Ataxia telangiectasia mutated (protein) |
| ATR | ATM–Rad3-related (protein) |
| BaP | Benzo(a)pyrene |
| BER | Base excision repair complex |
| CAT | Catalase, |
| CDK1 | Ciclin dependent kinase 1 |
| ε-A | 1N6 etheno-adenine |
| ε-C | 3N4 etheno-cytosine |
| ETC | Electron transport chain |
| FAD | Flavin adenine dinucleotide |
| FapyAde | 4,6-diamino-5-formamidopyrimidine |
| FapyGua | 2,6-diamino-4-hydroxy-5-formamido- pyrimidine. |
| GSH | Glutathione |
| GPX1 | Glutathione peroxidase 1 |
| GSTP1 | Glutathione S transferase P1-1 |
| HIF-1 | Hypoxia inducible factor - 1 |
| HNE | trans-4-hydroxy-2-nonenal (HNE) |
| H2O2 | Hydrogen peroxide. |
| HPV/HR-HPV | Human papillomavirus /high-risk human papillomavirus |
| LPO | Lipid peroxydes |
| MAPK/ERK | Mitogen-activated protein kinase/Extracellular signal-activated Kinase |
| M2PK | M2 pyruvate kinase |
| NAD/NADH | Nicotinamide adenine di nucleotide/reduced |
| •NO | Nitric oxide |
| iNOS | Inducible nitric oxide synthase |
| 8-oxoAde | 8-oxo-7,8-dihydroadenine |
| 8-oxo-Gua | 8-hydroxyguanine, 8-hydroxy-2’-deoxyguanonsine |
| 1O2 | Singlet oxygen |
| O2•− | Superoxide ion |
| RNS/ROS/RONS | Reactive nitrogen/oxygen/oxygen and nitrogen species |
| SESN | Sestrins |
| SESN2 | Sestrin 2 |
| SOD | Superoxide dismutase |
| Tg | 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol) |
| TGFβ-1 | Tumor growth factor β-1 |
| TopBP1 | DNA topoisomerase II beta-binding protein 1 |
| TRX | Thioredoxine reductase |
1. Introduction
2. Generation of RONS

2.1. Peroxisomal β-Oxidation
2.2. P450 Isoenzyme Activity
2.3. Inflammation
2.4. Ischemia–Reperfusion Stress
2.5. Signaling Pathways Activation
3. Biochemical Effects of RONS
3.1. Genetic Damage

3.2. Protein Damage
3.3. Modulation of Protein Function
4. The Interplay Between OS and HPV Carcinogenesis
4.1. OS and HPV Infection
4.2. OS and Viral DNA Replication

4.3. OS and Apoptosis Suppression in HPV-Expressing Cells
4.4. OS Cell Signaling and Metabolic Modulation of HPV Transformed Cells
4.5. Adaptation of Advanced Neoplastic Cells to OS
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- zur Hausen, H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin. Cancer Biol. 1999, 9, 405–411. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer. 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Harrison, D.G.; Widder, J.; Grumbach, I.; Chen, W.; Weber, M.; Searles, C. Endothelial mechano-transduction, nitric oxide and vascular inflammation. J. Intern. Med. 2006, 259, 351–363. [Google Scholar] [CrossRef]
- Darley-Usmar, V.; Halliwell, B. Blood radicals: Reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharm. Res. 1996, 13, 649–662. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Invest. Dermatol. 1994, 102, 671–675. [Google Scholar]
- Jurkiewicz, B.A.; Buettner, G.R. Ultraviolet light-induced free radical formation in skin: An electron paramagnetic resonance study. Photochem. Photobiol. 1994, 59, 1–4. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radia.t Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Breimer, L.H. Repair of DNA damage induced by reactive oxygen species. Free Radic. Res. Commun. 1991, 14, 159–171. [Google Scholar] [CrossRef]
- Ananthaswamy, H.N.; Pierceall, W.E. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem. Photobiol. 1990, 52, 1119–1136. [Google Scholar] [CrossRef]
- Danno, K.; Horio, T.; Takigawa, M.; Imamura, S. Role of oxygen intermediates in UV-induced epidermal cell injury. J. Invest. Dermatol. 1984, 83, 166–168. [Google Scholar] [CrossRef]
- Miyachi, Y. Photoaging from an oxidative standpoint. J. Dermatol. Sci. 1995, 9, 79–86. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol Rev. 2002, 82, 47–95. [Google Scholar]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–6142. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–1028. [Google Scholar]
- Lenaz, G.; Genova, M.L. Supramolecular organisation of the mitochondrial respiratory chain: A new challenge for the mechanism and control of oxidative phosphorylation. Adv. Exp. Med. Biol. 2012, 748, 107–144. [Google Scholar] [CrossRef]
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef]
- Di Mascio, P.; Briviba, K.; Bechara, E.J.; Medeiros, M.H.; Sies, H. Reaction of peroxynitrite and hydrogen peroxide to produce singlet molecular oxygen (1deltag). Methods Enzymol. 1996, 269, 395–400. [Google Scholar] [CrossRef]
- van Asbeck, B.S. Oxygen toxicity: Role of hydrogen peroxide and iron. Adv. Exp. Med. Biol. 1990, 264, 235–246. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef]
- Moriya, M.; Pandya, G.A.; Johnson, F.; Grollman, A.P. Cellular response to exocyclic DNA adducts. IARC Sci. Publ. 1999, 150, 263–270. [Google Scholar]
- Martínez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med. 2011, 51, 17–29. [Google Scholar] [CrossRef]
- Ascenzi, P.; di Masi, A.; Sciorati, C.; Clementi, E. Peroxynitrite-An ugly biofactor? Biofactors. 2010, 36, 264–273. [Google Scholar] [CrossRef]
- Dansen, T.B.; Wirtz, K.W. The peroxisome in oxidative stress. IUBMB Life 2001, 51, 223–230. [Google Scholar] [CrossRef]
- Kasai, H.; Okada, Y.; Nishimura, S.; Rao, M.S.; Reddy, J.K. Formation of 8-hydroxy deoxyguanosine in liver DNA of rats following long-term exposure to a peroxisome proliferator. Cancer Res. 1989, 49, 2603–2605. [Google Scholar]
- Oikawa, I.; Novikoff, P.M. Catalase-negative peroxisomes: Transient appearance in rat hepatocytes during liver regeneration after partial hepatectomy. Am. J. Pathol. 1995, 146, 673–687. [Google Scholar]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar]
- Goeptar, A.R.; Scheerens, H.; Vermulen, N.P. Oxygen and xenobiotic reductase activities of cytochrome P450. Crit. Rev. Toxicol. 1995, 25, 25–65. [Google Scholar]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992, 6, 724–730. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003, 4, 276–285. [Google Scholar] [CrossRef]
- Ohshima, H.; Bartsch, H. Chronic infections and inflammatory processes as cancer risk factors: Possible role of nitric oxide in carcinogenesis. Mutat. Res. 1994, 305, 253–264. [Google Scholar] [CrossRef]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: how, where, and what for? Free Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, C227–241. [Google Scholar]
- Kayyali, U.; Donaldson, C.; Huang, H.; Abdelnour, R.; Hassoun, P. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J. Biol. Chem. 2001, 276, 14359–14365. [Google Scholar]
- Plateel, M.; Dehouck, M.P.; Torpier, G.; Cecchelli, R.; Teissier, E. Hypoxia increases the susceptibility to oxidant stress and the permeability of the blood-brain barrier endothelial cell monolayer. J. Neurochem. 1995, 65, 2138–2145. [Google Scholar]
- Al-Mehdi, A.B; Zhao, G.; Dodia, C.; Tozawa, K; Costa, K.; Muzykantov, V.; Ross, C.; Blecha, F.; Dinauer, M.; Fisher, A.B. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res. 1998, 83, 730–737. [Google Scholar]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J. 2012, 31, 2448–60. [Google Scholar] [CrossRef]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef]
- Cadet, J.; Loft, S.; Olinski, R.; Evans, M.D.; Bialkowski, K.; Richard Wagner, J.; Dedon, P.C.; Møller, P.; Greenberg, M.M.; Cooke, M.S. Biologically relevant oxidants and terminology, classification and nomenclature of oxidatively generated damage to nucleobases and 2-deoxyribose in nucleic acids. Free Radic. Res. 2012, 46, 367–381. [Google Scholar] [CrossRef]
- Himmelstein, M.W.; Boogaard, P.J.; Cadet, J.; Farmer, P.B.; Kim, J.H.; Martin, E.A.; Persaud, R.; Shuker, D.E. Creating context for the use of DNA adduct data in cancer risk assessment: II. Overview of methods of identification and quantitation of DNA damage. Crit. Rev. Toxicol. 2009, 39, 679–694. [Google Scholar] [CrossRef]
- Pilger, A.; Rüdiger, H.W. 8-Hydroxy-2'-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int. Arch. Occup. Environ. Health 2006, 80, 1–15. [Google Scholar] [CrossRef]
- Revich, G.G.; Beattie, K.L. Utilization of 1,N6-etheno-2'-deoxyadenosine 5'-triphosphate during DNA synthesis on natural templates, catalyzed by DNA polymerase I of Escherichia coli. Carcinogenesis 1986, 7, 1569–1576. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. 2-Hydroxy-dATP is incorporated opposite G by Escherichia coli DNA polymerase III resulting in high mutagenicity. Nucleic Acids Res. 2000, 28, 1640–1646. [Google Scholar] [CrossRef]
- Levine, R.L.; Yang, I.Y.; Hossain, M.; Pandya, G.A.; Grollman, A.P.; Moriya, M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000, 60, 4098–4104. [Google Scholar]
- Pandya, G.A.; Moriya, M. 1,N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry 1996, 35, 11487114–11487192. [Google Scholar]
- Moriya, M.; Zhang, W.; Johnson, F.; Grollman, A.P. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proc. Natl. Acad. Sci. USA 1994, 91, 11899–11903. [Google Scholar]
- Esterbauer, H.; Eckl, P.; Ortner, A. Possible mutagens derived from lipids and lipid precursors. Mutat Res. 1990, 238, 223–233. [Google Scholar] [CrossRef]
- Tudek, B.; Winczura, A.; Janik, J.; Siomek, A.; Foksinski, M.; Oliński, R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am. J. Transl. Res. 2010, 2, 254–284. [Google Scholar]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. NY Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef]
- Butterfield, D.A. Oxidative stress in neurodegenerative disorders. Antioxid. Redox. Signal. 2006, 8, 1971–1973. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Keller, J.N. Antioxidants and antioxidant treatment in disease. Biochem. Biophys. Acta. 2012, 1822, 615. [Google Scholar] [CrossRef]
- England, K.; Cotter, T.G. Direct oxidative modifications of signaling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005, 10, 237–245. [Google Scholar] [CrossRef]
- De Marco, F.; Bucaj, E.; Foppoli, C.; Fiorini, A.; Blarzino, C.; Filipi, K.; Giorgi, A.; Schininà, M.E.; di Domenico, F.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Oxidative stress in HPV-Driven viral carcinogenesis: Redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One. 2012, 7, e34366. [Google Scholar]
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells. 2010, 30, 1–12. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Flohe, L.; Brigelius-Flohe, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox regulation of NF-kappa B activation. Free Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Aslund, F.; Zheng, M.; Beckwith, J.; Storz, G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. USA 1999, 96, 6161–6165. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Fernandez, C.; Sharrard, R.M.; Talbot, M.; Reed, B.D.; Monks, N. Evaluation of the significance of polyamines and their oxidases in the aetiology of human cervical carcinoma. Br. J. Cancer 1995, 72, 1194–1199. [Google Scholar] [CrossRef]
- Siegel, E.M.; Patel, N.; Lu, B.; Lee, J.H.; Nyitray, A.G.; Huang, X.; Villa, L.L.; Franco, E.L.; Giuliano, A.R. Circulating biomarkers of iron storage and clearance of incident human papillomavirus infection. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 859–865. [Google Scholar] [CrossRef]
- Safaeian, M.; Hildesheim, A.; Gonzalez, P.; Yu, K.; Porras, C.; Li, Q.; Rodriguez, A.C.; Sherman, M.E.; Schiffman, M.; Wacholder, S.; et al. Single nucleotide polymorphisms in the PRDX3 and RPS19 and risk of HPV persistence and cervical precancer/cancer. PLoS One 2012, 7, e33619. [Google Scholar]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J Virol. 2009, 83, 10515–10126. [Google Scholar] [CrossRef]
- Schmitt, J.; Schlehofer, J.R.; Mergener, K.; Gissmann, L.; zur Hausen, H. Amplification of bovine papillomavirus DNA by N-methyl-N'-nitro-N-nitrosoguanidine, ultraviolet irradiation, or infection with herpes simplex virus. Virology 1989, 172, 73–81. [Google Scholar]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A functional interaction between the HPV16 E2 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar]
- Achanta, G.; Huang, P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004, 64, 6233–6239. [Google Scholar]
- Giampieri, S.; Storey, A. Repair of UV-induced thymine dimers is compromised in cells expressing the E6 protein from human papillomaviruses types 5 and 18. Br. J. Cancer. 2004, 90, 2203–2209. [Google Scholar]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA damage signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef]
- Mincheva, A.; Gissmann, L.; zur Hausen, H. Chromosomal integration sites of human papillomavirus DNA in three cervical cancer cell lines mapped by in situ hybridization. Med. Microbiol. Immunol. 1987, 176, 245–256. [Google Scholar]
- Heller, A. Apoptosis-inducing high •NO concentrations are not sustained either in nascent or in developed cancers. Chem. Med. Chem. 2008, 3, 1493–1499. [Google Scholar]
- Goldstein, S.; Merényi, G. The chemistry of peroxynitrite: implications for biological activity. Methods Enzymol. 2008, 436, 49–61. [Google Scholar] [CrossRef]
- Dulak, J.; Józkowicz, A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox. Signal. 2003, 5, 123–132. [Google Scholar] [CrossRef]
- Morbidelli, L.; Donnini, S.; Ziche, M. Role of nitric oxide in tumor angiogenesis. Cancer Treat. Res. 2004, 117, 155–167. [Google Scholar] [CrossRef]
- Mazibrada, J.; Rittà, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef]
- De Marco, F.; Bucaj, E.; Foppoli, C.; Fiorini, A.; Blarzino, C.; Filipi, K.; Giorgi, A.; Schininà, M.E.; Di Domenico, F.; Coccia, R.; et al. Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One 2012, 7, e34366. [Google Scholar]
- Vodovot, Y.; Bogdan, C.; Paik, J.; Xie, Q.W.; Nathan, C. Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J. Exp. Med. 1993, 178, 605–613. [Google Scholar] [CrossRef]
- Mitani, T.; Terashima, M.; Yoshimura, H.; Nariai, Y.; Tanigawa, Y. TGF-β1 enhances degradation of IFN-γ-induced iNOS protein via proteasomes in RAW 264.7 cells. Nitric. Oxide 2005, 13, 78–87. [Google Scholar] [CrossRef]
- Baritaki, S.; Sifakis, S.; Huerta-Yepez, S.; Neonakis, I.K.; Soufla, G.; Bonavida, B.; Spandidos, D.A. Overexpression of VEGF and TGF-beta1 mRNA in Pap smears correlates with progression of cervical intraepithelial neoplasia to cancer: Implication of YY1 in cervical tumorigenesis and HPV infection. Int. J. Oncol. 2007, 31, 69–79. [Google Scholar]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 8, 355–360. [Google Scholar]
- López-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar]
- Butz, K.; Shahabeddin, L.; Geise, C.; Spitkovsky, D.; Ullmann, A.; Hoppe-Seyler, F. Functional p53 protein in human papillomavirus-positive cancer cells. Oncogene 1995, 10, 927–933. [Google Scholar]
- Butz, K.; Whitaker, N.; Denk, C.; Ullmann, A.; Geisen, C.; Hoppe-Seyler, F. Induction of the p53-target gene GADD45 in HPV-positive cancer cells. Oncogene 1999, 18, 2381–2386. [Google Scholar]
- Ding, B.; Chi, S.G.; Kim, S.H.; Kang, S.; Cho, J.H.; Kim, D.S.; Cho, N.H. Role of p53 in antioxidant defence of HPV-positive cervical carcinoma cells following H2O2 exposure. J. Cell. Sci. 2007, 120, 2284–2294. [Google Scholar] [CrossRef]
- De Marco, F.; Marcante, M.L. HPV-16 E6-E7 differential transcription induced in Siha cervical cancer cell line by interferons. J. Biol. Regul. Homeost. Agents 1993, 7, 15–21. [Google Scholar]
- Abdulkarim, B.; Sabri, S.; Deutsch, E.; Chagraoui, H.; Maggiorella, L.; Thierry, J.; Eschwege, F.; Vainchenker, W.; Chouaïb, S.; Bourhis, J. Antiviral agent Cidofovir restores p53 function and enhances the radio-sensitivity in HPV-associated cancers. Oncogene 2002, 21, 2334–2346. [Google Scholar] [CrossRef]
- Divya, C.S.; Pillai, M.R. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog. 2006, 45, 320–332. [Google Scholar] [CrossRef]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin A induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef]
- De Marco, F.; Perluigi, M.; Foppoli, C.; Blarzino, C.; Cini, C.; Coccia, R.; Venuti, A. UVB irradiation down-regulates HPV-16 RNA expression: Implications for malignant progression of transformed cells. Virus Res. 2007, 130, 249–259. [Google Scholar] [CrossRef]
- Mouret, S.; Sauvaigo, S.; Peinnequin, A.; Favier, A.; Beani, J.C.; Leccia, M.T. E6* oncoprotein expression of human papillomavirus type-16 determines different ultraviolet sensitivity related to glutathione and glutathione peroxidase antioxidant defence. Exp. Dermatol. 2005, 14, 401–410. [Google Scholar] [CrossRef]
- Shim, J.H.; Kim, K.H.; Cho, Y.S.; Choi, H.S.; Song, E.Y.; Myung, P.K.; Kang, J.S.; Suh, S.K.; Park, S.N.; Yoon, D.Y. Protective effect of oxidative stress in HaCaT keratinocytes expressing E7 oncogene. Amino Acids. 2008, 34, 135–141. [Google Scholar]
- Mileo, A.M.; Abbruzzese, C.; Mattarocci, S.; Bellacchio, E.; Pisano, P.; Federico, A.; Maresca, V.; Picardo, M.; Giorgi, A.; Maras, B.; Schininà, M.E.; Paggi, M.G. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One 2009, 13, e7254. [Google Scholar]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef]
- Rösl, F.; Das, B.C.; Lengert, M.; Geletneky, K.; zur Hausen, H. Antioxidant-induced changes of the AP-1 transcription complex are paralleled by a selective suppression of human papillomavirus transcription. J. Virol. 1997, 71, 362–370. [Google Scholar]
- Soto, U.; Denk, C.; Finzer, P.; Hutter, K.J.; zur Hausen, H.; Rösl, F. Genetic complementation to non-tumorigenicity in cervical-carcinoma cells correlates with alterations in AP-1 composition. Int. J. Cancer. 2000, 86, 811–817. [Google Scholar] [CrossRef]
- Soto, U.; Das, B.C.; Lengert, M.; Finzer, P.; zur Hausen, H.; Rösl, F. Conversion of HPV 18 positive non-tumorigenic HeLa-fibroblast hybrids to invasive growth involves loss of TNF-alpha mediated repression of viral transcription and modification of the AP-1 transcription complex. Oncogene 1999, 18, 3187–3198. [Google Scholar] [CrossRef]
- Prusty, B.K.; Das, B.C. Constitutive activation of transcription factor AP-1 in cervical cancer and suppression of human papillomavirus (HPV) transcription and AP-1 activity in HeLa cells by curcumin. Int. J. Cancer. 2005, 113, 951–960. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Dürr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 1291–1296. [Google Scholar]
- Mazurek, S.; Zwerschke, W.; Jansen-Dürr, P.; Eigenbrodt, E. Effects of the human papilloma virus HPV-16 E7 oncoprotein on glycolysis and glutaminolysis: Role of pyruvate kinase type M2 and the glycolytic-enzyme complex. Biochem. J. 2001, 356, 247–256. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef]
- zur Hausen, H. Human papilloma viruses and their possible role in squamous cell carcinomas. Curr. Top. Microbiol. Immunol. 1977, 78, 1–30. [Google Scholar] [CrossRef]
- Slater, T.F.; Bajardi, F.; Benedetto, C.; Bussolati, G.; Cianfano, S.; Dianzani, M.U.; Ghiringhello, B.; Nöhammer, G.; Rojanapo, W.; Schauenstein, E. Protein thiols in normal and neoplastic human uterine cervix. FEBS Lett. 1985, 187, 267–271. [Google Scholar]
- Kwasniewska, A.; Gozdzicka-Józefiak, A.; Borzecki, A.; Baranowski, W. DNA adducts in squamous cell cervical carcinomas associated with HPV infection. Eur. J. Gynaec. Oncol. 2003, 3, 359–361. [Google Scholar]
- Sgambato, A.; Zannoni, G.F.; Faraglia, B.; Camerini, A.; Tarquini, E.; Spada, D.; Cittadini, A. Decreased expression of the CDK inhibitor p27Kip1 and increased oxidative DNA damage in the multistep process of cervical carcinogenesis. Gynecol. Oncol. 2004, 92, 776–783. [Google Scholar] [CrossRef]
- Hedley, D.; Pintilie, M.; Woo, J.; Nicklee, T.; Morrison, A.; Birle, D.; Fyles, A.; Milosevic, M.; Hill, R. Up-regulation of the redox mediators thioredoxin and apurinic/apyrimidinic excision (APE)/Ref-1 in hypoxic microregions of invasive cervical carcinomas, mapped using multispectral, wide-field fluorescence image analysis. Am. J. Pathol. 2004, 164, 557–565. [Google Scholar] [CrossRef]
- Del Nonno, F.; Pisani, G.; Visca, P.; Signore, F.; Grillo, L.R.; Baiocchini, A.; Garbuglia, A.R.; Sepe, S.; Piacentini, M.; Falasca, L. Role and predictive strength of transglutaminase type 2 expression in premalignant lesions of the cervix. Mod. Pathol. 2011, 24, 855–865. [Google Scholar] [CrossRef]
- Li, L.Q.; Chen, C.L.; Cao, Z.Y.; Liao, Q.P.; Du, H.J.; Zhan, S.B.; Zhou, L.; Zeng, Y. Expression of peroxiredoxin III in cervical lesions. ZhonghuaShi Yan He Lin Chuang Bing Du Xue Za Zhi 2009, 23, 443–445. [Google Scholar]
- Perluigi, M.; Giorgi, A.; Blarzino, C.; De Marco, F.; Foppoli, C.; Di Domenico, F.; Butterfield, D.A.; Schininà, M.E.; Cini, C.; Coccia, R. Proteomics analysis of protein expression and specific protein oxidation in human papillomavirus transformed keratinocytes upon UVB irradiation. J. Cell Mol. Med. 2009, 13, 1809–1822. [Google Scholar] [CrossRef]
- Carrero, Y.; Callejas, D.; Alaña, F.; Silva, C.; Mindiola, R.; Mosquera, J. Increased vascular endothelial growth factor expression, CD3-positive cell infiltration, and oxidative stress in premalignant lesions of the cervix. Cancer 2009, 115, 3680–3688. [Google Scholar] [CrossRef]
- Maldonado, P.; Negrini, L.A.; Kaizer, R.R.; Zanin, R.F.; Araújo Mdo, C.; Battisti, V.; Morsch, V.M.; Schetinger, M.R. Oxidative status in patients submitted to conization and radiation treatments for uterine cervix neoplasia. Clin. Chim. Acta. 2006, 366, 174–178. [Google Scholar] [CrossRef]
- Sharma, A.; Rajappa, M.; Satyam, A.; Sharma, M. Oxidant/anti-oxidant dynamics in patients with advanced cervical cancer: correlation with treatment response. Mol. Cell. Biochem. 2010, 341, 65–72. [Google Scholar] [CrossRef]
- Weitmann, H.D.; Gustorff, B.; Vaupel, P.; Knocke, T.H.; Pötter, R. Oxygenation status of cervical carcinomas before and during spinal anesthesia for application of brachytherapy. Strahlenther Onkol. 2003, 179, 633–640. [Google Scholar] [CrossRef]
- Ho, G.Y.; Kadish, A.S.; Burk, R.D.; Basu, J.; Palan, P.R.; Mikhail, M.; Romney, S.L. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int. J. Cancer 1998, 78, 281–285. [Google Scholar]
- Ho, G.Y.; Palan, P.R.; Basu, J.; Romney, S.L.; Kadish, A.S.; Mikhail, M.; Wassertheil-Smoller, S.; Runowicz, C.; Burk, R.D. Viral characteristics of human papillomavirus infection and antioxidant levels as risk factors for cervical dysplasia. Int. J. Cancer. 1998, 78, 594–599. [Google Scholar] [CrossRef]
- Alam, S.; Conway, M.J.; Chen, H.S.; Meyers, C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J. Virol. 2008, 82, 1053–1058. [Google Scholar] [CrossRef]
- Reddy, V.G.; Khanna, N.; Singh, N. Vitamin C augments chemotherapeutic response of cervical carcinoma HeLa cells by stabilizing P53. Biochem. Biophys. Res. Commun. 2001, 282, 409–415. [Google Scholar] [CrossRef]
- López, J.; Poitevin, A.; Mendoza-Martínez, V.; Pérez-Plasencia, C.; García-Carrancá, A. Cancer-initiating cells derived from established cervical cell lines exhibit stem-cell markers and increased radioresistance. BMC Cancer 2012, 28, 12–48. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Marco, F. Oxidative Stress and HPV Carcinogenesis. Viruses 2013, 5, 708-731. https://doi.org/10.3390/v5020708
De Marco F. Oxidative Stress and HPV Carcinogenesis. Viruses. 2013; 5(2):708-731. https://doi.org/10.3390/v5020708
Chicago/Turabian StyleDe Marco, Federico. 2013. "Oxidative Stress and HPV Carcinogenesis" Viruses 5, no. 2: 708-731. https://doi.org/10.3390/v5020708
APA StyleDe Marco, F. (2013). Oxidative Stress and HPV Carcinogenesis. Viruses, 5(2), 708-731. https://doi.org/10.3390/v5020708