Directional Spread of Alphaherpesviruses in the Nervous System

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Alphaherpesvirinae
1.2. Model Alphaherpesviruses
1.3. Neuronal Architecture, Directional Spread of Infection and Alphaherpesvirus Pathogenesis

2. The Alphaherpesvirus Infectious Cycle
2.1. Virion Attachment and Entry

2.2. Genome Replication and Nuclear Egress
2.3. Virion Assembly, Secondary Envelopment and Cellular Egress
3. Utilization of the Host Cytoskeleton for Directional Spread of Infection in Neurons
3.1. Viral Particle Movement Requires Active Transport
3.2. The Actin and Microtubule Cytoskeletons
3.3. Retrograde Transport During Viral Entry
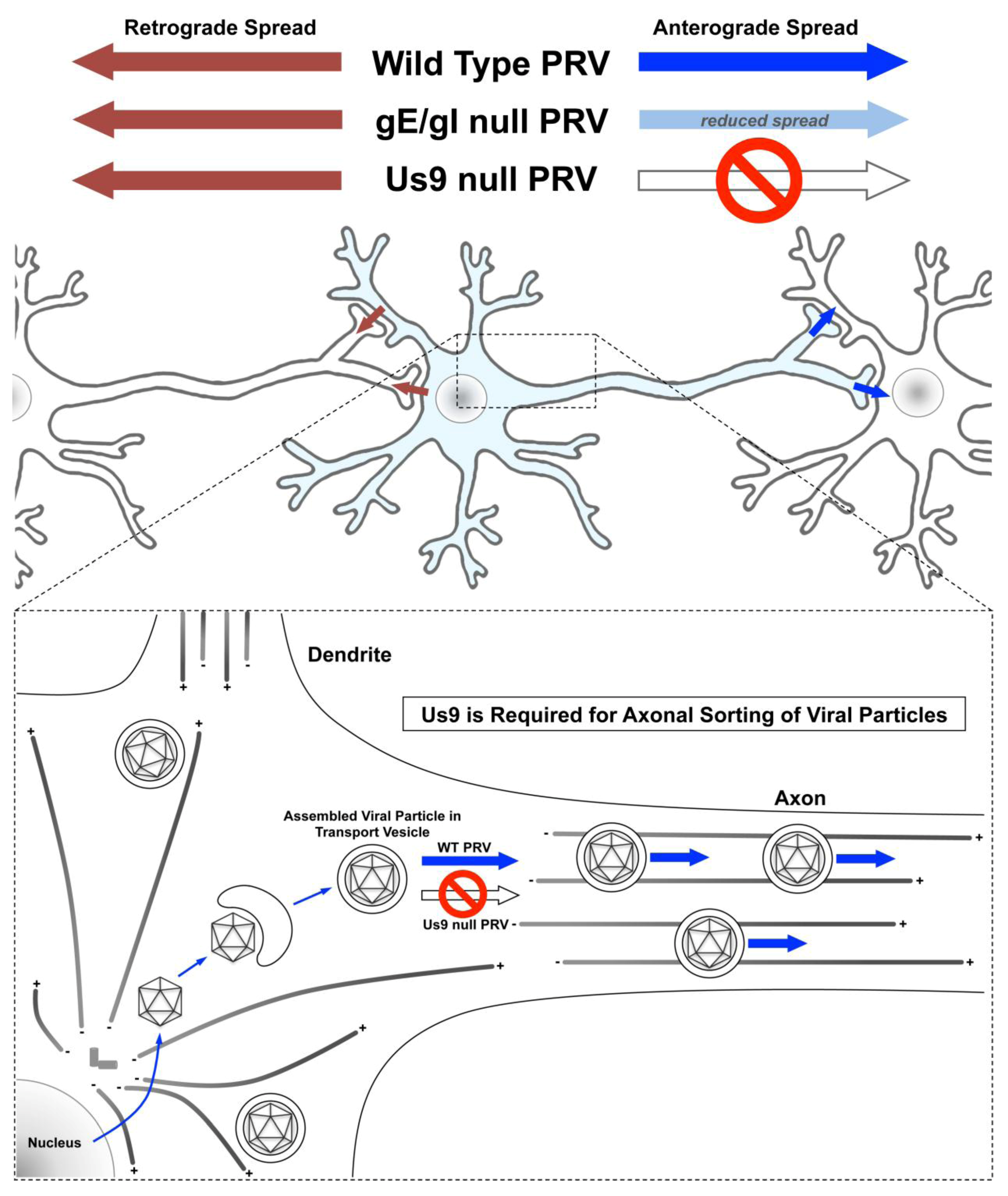
3.4. Anterograde Viral Transport and Axonal Sorting

3.5. Reorganization of the Cytoskeleton and Alteration of Organelle Dynamics During Infection
4. Concluding Remarks
Conflict of Interest
Acknowledgements
References
- Pellett, P.E.; Roizman, B. The Family: Herpesviridae a Brief Introduction. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott, Williams, and Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2479–2500. [Google Scholar]
- Goodpasture, E.W.; Teague, O. Transmission of the virus of herpes febrilis along nerves in experimentally infected rabbits. J. Med. Res. 1923, 44, 139–184. [Google Scholar]
- Goodpasture, E.W.; Teague, O. Experimental production of herpetic lesions in organs and tissues of the rabbit. J. Med. Res. 1923, 44, 121–138. [Google Scholar]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef]
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L.W. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Patho. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef]
- Smith, G.; Enquist, L.W. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4873–4878. [Google Scholar]
- Szpara, M.L.; Tafuri, Y.R.; Enquist, L.W. Preparation of viral DNA from nucleocapsids. J. Vis. Exp. 2011. [Google Scholar]
- Ekstrand, M.I.; Enquist, L.W.; Pomeranz, L.E. The alpha-herpesviruses: Molecular pathfinders in nervous system circuits. Trends Mol. Med. 2008, 14, 134–140. [Google Scholar]
- Antinone, S.; Smith, G. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2009, 84, 1504–1512. [Google Scholar] [CrossRef]
- Smith, G. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Kimberlin, D.W. Herpes simplex virus infections in neonates and early childhood. Semin. Pediatr. Infect. Dis 2005, 16, 271–281. [Google Scholar] [CrossRef]
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221. [Google Scholar]
- Wald, A.; Zeh, J.; Selke, S.; Warren, T.; Ryncarz, A.J.; Ashley, R.; Krieger, J.N.; Corey, L. Reactivation of genital herpes simplex virus type 2 infection in asymptomatic seropositive persons. N Engl. J. Med. 2000, 342, 844–850. [Google Scholar] [CrossRef]
- Heininger, U.; Seward, J.F. Varicella. Lancet 2006, 368, 1365–1376. [Google Scholar] [CrossRef]
- Kost, R.G.; Straus, S.E. Postherpetic neuralgia—pathogenesis, treatment, and prevention. N. Engl. J. Med. 1996, 335, 32–42. [Google Scholar] [CrossRef]
- Rasband, M.N. The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 2010, 11, 552–562. [Google Scholar] [CrossRef]
- Moughamian, A.J.; Holzbaur, E.L.F. Synaptic vesicle distribution by conveyor belt. Cell 2012, 148, 849–851. [Google Scholar] [CrossRef]
- Enquist, L.W. Five questions about viral trafficking in neurons. PLoS Pathog. 2012, 8, e1002472. [Google Scholar]
- Zaichick, S.V.; Bohannon, K.P.; Smith, G.A. Alphaherpesviruses and the cytoskeleton in neuronal infections. Viruses 2011, 3, 941–981. [Google Scholar] [CrossRef]
- Topp, K.S.; Meade, L.B.; LaVail, J.H. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: Relevance for the retrograde transport of herpes simplex virus. J. Neurosci. 1994, 14, 318–325. [Google Scholar]
- Chen, S.-H.; Yao, H.-W.; Huang, W.-Y.; Hsu, K.-S.; Lei, H.-Y.; Shiau, A.-L.; Chen, S.-H. Efficient reactivation of latent herpes simplex virus from mouse central nervous system tissues. J. Virol. 2006, 80, 12387–12392. [Google Scholar] [CrossRef]
- Tyler, K.L. Herpes simplex virus infections of the central nervous system: Encephalitis and meningitis, including mollaret's. Herpes 2004, 11 Suppl. 2, 57–64. [Google Scholar]
- Smith, C.; Lachmann, R.H.; Efstathiou, S. Expression from the herpes simplex virus type 1 latency-associated promoter in the murine central nervous system. J. Gen. Virol. 2000, 81, 649–662. [Google Scholar]
- Cabrera, C.V.; Wohlenberg, C.; Openshaw, H.; Rey-Mendez, M.; Puga, A.; Notkins, A.L. Herpes simplex virus DNA sequences in the cns of latently infected mice. Nature News 1980, 288, 288–290. [Google Scholar] [CrossRef]
- Fraser, N.W.; Lawrence, W.C.; Wroblewska, Z.; Gilden, D.H.; Koprowski, H. Herpes simplex type 1 DNA in human brain tissue. PNAS 1981, 78, 6461–6465. [Google Scholar]
- Marsden, H. Herpes simplex virus in latent infection. Nature News 1980, 228, 212–213. [Google Scholar] [CrossRef]
- Rock, D.L.; Fraser, N.W. Detection of hsv-1 genome in central nervous system of latently infected mice. Nature News 1983, 302, 523–525. [Google Scholar] [CrossRef]
- Sekizawa, T.; Openshaw, H. Encephalitis resulting from reactivation of latent herpes simplex virus in mice. J. Virol. 1984, 50, 263–266. [Google Scholar]
- Sequiera, L.W.; Jennings, L.C.; Carrasco, L.H.; Lord, M.A.; Curry, A.; Sutton, R.N. Detection of herpes-simplex viral genome in brain tissue. Lancet 1979, 2, 609–612. [Google Scholar]
- Sabó, A.; Rajcáni, J. Latent pseudorabies virus infection in pigs. Acta. virologica 1976, 20, 208–214. [Google Scholar]
- Beran, G.W.; Davies, E.B.; Arambulo, P.V.; Will, L.A.; Hill, H.T.; Rock, D.L. Persistence of pseudorabies virus in infected swine. J. Am. Vet. Med. Assoc. 1980, 176, 998–1000. [Google Scholar]
- Wheeler, J.G.; Osorio, F.A. Investigation of sites of pseudorabies virus latency, using polymerase chain reaction. Am. J. Vet. Res. 1991, 52, 1799–1803. [Google Scholar]
- Tham, K.M.; Motha, M.X.; Horner, G.W.; Ralston, J.C. Polymerase chain reaction amplification of latent aujeszky's disease virus in dexamethasone treated pigs. Arch. Virol. 1994, 136, 197–205. [Google Scholar] [CrossRef]
- Capua, I.; Fico, R.; Banks, M.; Tamba, M.; Calzetta, G. Isolation and characterisation of an aujeszky's disease virus naturally infecting a wild boar (sus scrofa). Vet. Microbiol. 1997, 55, 141–146. [Google Scholar] [CrossRef]
- Brittle, E.E.; Reynolds, A.E.; Enquist, L.W. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J. Virol. 2004, 78, 12951–12963. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Pathogenesis of neurotropic herpesviruses: Role of viral glycoproteins in neuroinvasion and transneuronal spread. Virus Res. 2003, 92, 197–206. [Google Scholar] [CrossRef]
- Esiri, M.M. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J. Neurol. Sci. 1982, 54, 209–226. [Google Scholar] [CrossRef]
- Casrouge, A.; Zhang, S.-Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human unc-93b deficiency. Science 2006, 314, 308–312. [Google Scholar]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. Unc93b1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. [Google Scholar]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to hsv-1 in human ipsc-derived tlr3-deficient cns cells. Nature 2012, 491, 769–773. [Google Scholar]
- Conrady, C.D.; Drevets, D.A.; Carr, D.J.J. Herpes simplex type i (hsv-1) infection of the nervous system: Is an immune response a good thing? J. Neuroimmunol. 2010, 220, 1–9. [Google Scholar] [CrossRef]
- Pérez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human traf3 adaptor molecule deficiency leads to impaired toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef]
- Zhou, Y.; Ye, L.; Wan, Q.; Zhou, L.; Wang, X.; Li, J.; Hu, S.; Zhou, D.; Ho, W. Activation of toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J. Neurosci. Res. 2009, 87, 2916–2925. [Google Scholar] [CrossRef]
- Reinert, L.S.; Harder, L.; Holm, C.K.; Iversen, M.B.; Horan, K.A.; Dagnæs-Hansen, F.; Ulhøi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. Tlr3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of cns infection in mice. J. Clin. Invest. 2012, 122, 1368–1376. [Google Scholar]
- Mcgavern, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011, 11, 318–329. [Google Scholar]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar]
- Mettenleiter, T.; Zsak, L.; Zuckermann, F.; Sugg, N.; Kern, H.; Ben-Porat, T. Interaction of glycoprotein giii with a cellular heparinlike substance mediates adsorption of pseudorabies virus. J. Virol. 1990, 64, 278–286. [Google Scholar]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein c-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein b. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins b and c, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the tnf/ngf receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-o-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grünewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar]
- Peeters, B.; Pol, J.; Gielkens, A.; Moormann, R. Envelope glycoprotein gp50 of pseudorabies virus is essential for virus entry but is not required for viral spread in mice. J. Virol. 1993, 67, 170–177. [Google Scholar]
- Peeters, B.; de Wind, N.; Hooisma, M.; Wagenaar, F.; Gielkens, A.; Moormann, R. Pseudorabies virus envelope glycoproteins gp50 and gii are essential for virus penetration, but only gii is involved in membrane fusion. J. Virol. 1992, 66, 894–905. [Google Scholar]
- Ch'ng, T.H.; Spear, P.G.; Struyf, F.; Enquist, L.W. Glycoprotein d-independent spread of pseudorabies virus infection in cultured peripheral nervous system neurons in a compartmented system. J. Virol. 2007, 81, 10742–10757. [Google Scholar] [CrossRef]
- Connolly, S.A.; Whitbeck, J.J.; Rux, A.H.; Krummenacher, C.; van Drunen Littel-van den Hurk, S.; Cohen, G.H.; Eisenberg, R.J. Glycoprotein d homologs in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human hvec(nectin-1) with different affinities. Virology 2001, 280, 7–18. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Jogger, C.R.; Spear, P.G. Cellular expression of alphaherpesvirus gd interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gd receptor. Virology 2000, 268, 147–158. [Google Scholar] [CrossRef]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 866–871. [Google Scholar]
- Berarducci, B.; Rajamani, J.; Reichelt, M.; Sommer, M.; Zerboni, L.; Arvin, A.M. Deletion of the first cysteine-rich region of the varicella-zoster virus glycoprotein e ectodomain abolishes the ge and gi interaction and differentially affects cell-cell spread and viral entry. J. Virol. 2009, 83, 228–240. [Google Scholar] [CrossRef]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 2006, 127, 305–316. [Google Scholar]
- Li, Q.; Krogmann, T.; Ali, M.A.; Tang, W.-J.; Cohen, J.I. The amino terminus of varicella-zoster virus (vzv) glycoprotein e is required for binding to insulin-degrading enzyme, a vzv receptor. J. Virol. 2007, 81, 8525–8532. [Google Scholar] [CrossRef]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gh-gl. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein b from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar]
- Granzow, H.; Klupp, B.G.; Mettenleiter, T. Entry of pseudorabies virus: An immunogold-labeling study. J. Virol. 2005, 79, 3200–3205. [Google Scholar] [CrossRef]
- Luxton, G.W.G.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathogens 2010, 6, e1000991. [Google Scholar] [CrossRef]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes simplex virus replication: Roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 2009, 83, 1660–1668. [Google Scholar] [CrossRef]
- Coller, K.E.; Smith, G. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 2008, 9, 1458–1470. [Google Scholar] [CrossRef]
- Luxton, G.W.G.; Lee, J.I.-H.; Haverlock-Moyns, S.; Schober, J.M.; Smith, G. The pseudorabies virus vp1/2 tegument protein is required for intracellular capsid transport. J. Virol. 2006, 80, 201–209. [Google Scholar] [CrossRef]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of vp1–2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef]
- Ihara, S.; Feldman, L.; Watanabe, S.; Ben-Porat, T. Characterization of the immediate-early functions of pseudorabies virus. Virology 1983, 131, 437–454. [Google Scholar] [CrossRef]
- Kwong, A.D.; Frenkel, N. The herpes simplex virus virion host shutoff function. J. Virol. 1989, 63, 4834–4839. [Google Scholar]
- Ladin, B.F.; Blankenship, M.L.; Ben-Porat, T. Replication of herpesvirus DNA. V. Maturation of concatemeric DNA of pseudorabies virus to genome length is related to capsid formation. J. Virol. 1980, 33, 1151–1164. [Google Scholar]
- Mettenleiter, T.C. Herpesvirus assembly and egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar]
- Granzow, H.; Weiland, F.; Jöns, A.; Klupp, B.G.; Karger, A.; Mettenleiter, T. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: A reassessment. J. Virol. 1997, 71, 2072–2082. [Google Scholar]
- Lycke, E.; Hamark, B.; Johansson, M.; Krotochwil, A.; Lycke, J.; Svennerholm, B. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch. Virol. 1988, 101, 87–104. [Google Scholar] [CrossRef]
- Speese, S.D.; Ashley, J.; Jokhi, V.; Nunnari, J.; Barria, R.; Li, Y.; Ataman, B.; Koon, A.; Chang, Y.-T.; Li, Q.; et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic wnt signaling. Cell 2012, 149, 832–846. [Google Scholar] [CrossRef]
- Chang, Y.E.; Van Sant, C.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the u(l)31 gene of herpes simplex virus 1: Construction and phenotype in infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 ul31, ul34, and us3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. U(l)31 and u(l)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 u(l)34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef]
- Chang, Y.E.; Roizman, B. The product of the ul31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J. Virol. 1993, 67, 6348–6356. [Google Scholar]
- Mou, F.; Forest, T.; Baines, J.D. Us3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin a/c in infected cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef]
- Naldinho-Souto, R.; Browne, H.; Minson, T. Herpes simplex virus tegument protein vp16 is a component of primary enveloped virions. J. Virol. 2006, 80, 2582–2584. [Google Scholar] [CrossRef]
- Padula, M.E.; Sydnor, M.L.; Wilson, D.W. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 2009, 83, 4757–4765. [Google Scholar] [CrossRef]
- Baines, J.D.; Jacob, R.J.; Simmerman, L.; Roizman, B. The herpes simplex virus 1 ul11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 1995, 69, 825–833. [Google Scholar]
- Read, G.S.; Patterson, M. Packaging of the virion host shutoff (vhs) protein of herpes simplex virus: Two forms of the vhs polypeptide are associated with intranuclear b and c capsids, but only one is associated with enveloped virions. J. Virol. 2007, 81, 1148–1161. [Google Scholar] [CrossRef]
- McMillan, T.N.; Johnson, D.C. Cytoplasmic domain of herpes simplex virus ge causes accumulation in the trans-golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 2001, 75, 1928–1940. [Google Scholar] [CrossRef]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef]
- Wisner, T.W.; Johnson, D.C. Redistribution of cellular and herpes simplex virus proteins from the trans-golgi network to cell junctions without enveloped capsids. J. Virol. 2004, 78, 11519–11535. [Google Scholar] [CrossRef]
- Turcotte, S.; Letellier, J.; Lippé, R. Herpes simplex virus type 1 capsids transit by the trans-golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 2005, 79, 8847–8860. [Google Scholar] [CrossRef]
- Campadelli, G.; Brandimarti, R.; Di Lazzaro, C.; Ward, P.L.; Roizman, B.; Torrisi, M.R. Fragmentation and dispersal of golgi proteins and redistribution of glycoproteins and glycolipids processed through the golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1993, 90, 2798–2802. [Google Scholar]
- Kratchmarov, R.; Taylor, M.P.; Enquist, L.W. Making the case: Married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2012, 22, 1–12. [Google Scholar] [CrossRef]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382. [Google Scholar] [CrossRef]
- Taylor, M.P.; Kramer, T.; Lyman, M.G.; Kratchmarov, R.; Enquist, L.W. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. MBio 2012, 3, e00063-12. [Google Scholar]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef]
- Huang, J.; Lazear, H.M.; Friedman, H.M. Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 2011, 409, 12–16. [Google Scholar] [CrossRef]
- Antinone, S.E.; Zaichick, S.V.; Smith, G. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010. [Google Scholar]
- Negatsch, A.; Granzow, H.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 2010, 84, 13031–13035. [Google Scholar]
- Maresch, C.; Granzow, H.; Negatsch, A.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural analysis of virion formation and anterograde intraaxonal transport of the alphaherpesvirus pseudorabies virus in primary neurons. J. Virol. 2010, 84, 5528–5539. [Google Scholar]
- Lyman, M.; Feierbach, B.; Curanovic, D.; Bisher, M.; Enquist, L.W. Pseudorabies virus us9 directs axonal sorting of viral capsids. J. Virol. 2007, 81, 11363–11371. [Google Scholar]
- Feierbach, B.; Bisher, M.; Goodhouse, J.; Enquist, L.W. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. J. Virol. 2007, 81, 6846–6857. [Google Scholar] [CrossRef]
- Antinone, S.E.; Smith, G. Two modes of herpesvirus trafficking in neurons: Membrane acquisition directs motion. J. Virol. 2006, 80, 11235–11240. [Google Scholar] [CrossRef]
- Ch'ng, T.H.; Enquist, L.W. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein e. J. Virol. 2005, 79, 8835–8846. [Google Scholar] [CrossRef]
- del Rio, T.; Ch'ng, T.H.; Flood, E.A.; Gross, S.P.; Enquist, L.W. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 2005, 79, 3903–3919. [Google Scholar]
- Ohara, P.T.; Chin, M.S.; LaVail, J.H. The spread of herpes simplex virus type 1 from trigeminal neurons to the murine cornea: An immunoelectron microscopy study. J. Virol. 2000, 74, 4776–4786. [Google Scholar] [CrossRef]
- LaVail, J.H.; Topp, K.S.; Giblin, P.A.; Garner, J.A. Factors that contribute to the transneuronal spread of herpes simplex virus. J. Neurosci. Res. 1997, 49, 485–496. [Google Scholar] [CrossRef]
- Kristensson, K.; Sheppard, R.D.; Bornstein, M.B. Observations on uptake of herpes simplex virus in organized cultures of mammalian nervous tissue. Acta. Neuropathol. 1974, 28, 37–44. [Google Scholar] [CrossRef]
- Cook, M.L.; Stevens, J.G. Pathogenesis of herpetic neuritis and ganglionitis in mice: Evidence for intra-axonal transport of infection. Infect. Immun. 1973, 7, 272–288. [Google Scholar]
- Yamamoto, T.; Otani, S.; Shiraki, H. Ultrastructure of herpes simplex virus infection of the nervous system of mice. Acta. Neuropathol. 1973, 26, 285–299. [Google Scholar] [CrossRef]
- Hill, T.J.; Field, H.J.; Roome, A.P. Intra-axonal location of herpes simplex virus particles. J. Gen. Virol. 1972, 15, 233–235. [Google Scholar]
- Ibiricu, I.; Huiskonen, J.T.; Döhner, K.; Bradke, F.; Sodeik, B.; Grünewald, K. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011, 7, e1002406. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Boadle, R.A.; Aggarwal, A.; Tijono, B.; Rixon, F.J.; Diefenbach, R.J.; Cunningham, A.L. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 2009, 83, 3187–3199. [Google Scholar] [CrossRef]
- Snyder, A.; Polcicova, K.; Johnson, D.C. Herpes simplex virus ge/gi and us9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 2008, 82, 10613–10624. [Google Scholar] [CrossRef]
- Saksena, M.M.; Wakisaka, H.; Tijono, B.; Boadle, R.A.; Rixon, F.; Takahashi, H.; Cunningham, A.L. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 2006, 80, 3592–3606. [Google Scholar]
- Snyder, A.; Wisner, T.W.; Johnson, D.C. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 2006, 80, 11165–11177. [Google Scholar] [CrossRef]
- Tomishima, M.J.; Enquist, L.W. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 2001, 154, 741–752. [Google Scholar]
- Miranda-Saksena, M.; Armati, P.; Boadle, R.A.; Holland, D.J.; Cunningham, A.L. Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J. Virol. 2000, 74, 1827–1839. [Google Scholar] [CrossRef]
- Holland, D.J.; Miranda-Saksena, M.; Boadle, R.A.; Armati, P.; Cunningham, A.L. Anterograde transport of herpes simplex virus proteins in axons of peripheral human fetal neurons: An immunoelectron microscopy study. J. Virol. 1999, 73, 8503–8511. [Google Scholar]
- Penfold, M.E.; Armati, P.J.; Cunningham, A.L. Axonal transport of herpes simplex virions to epidermal cells: Evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 1994, 91, 6529–6533. [Google Scholar] [CrossRef]
- Curanovic, D.; Enquist, L.W. Directional transneuronal spread of alpha-herpesvirus infection. Future Virol. 2009, 4, 591. [Google Scholar]
- Curanovic, D.; Enquist, L.W. Virion-incorporated glycoprotein b mediates transneuronal spread of pseudorabies virus. J. Virol. 2009, 83, 7796–7804. [Google Scholar] [CrossRef]
- Cai, H.; Reinisch, K.; Ferro-Novick, S. Coats, tethers, rabs, and snares work together to mediate the intracellular destination of a transport vesicle. Dev. Cell. 2007, 12, 671–682. [Google Scholar] [CrossRef]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe. 2012, 12, 806–814. [Google Scholar] [CrossRef]
- Sodeik, B. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 2000, 8, 465–472. [Google Scholar] [CrossRef]
- Lyman, M.G.; Enquist, L.W. Herpesvirus interactions with the host cytoskeleton. J. Virol. 2009, 83, 2058–2066. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Enquist, L.W.; Feierbach, B. Actin and rho gtpases in herpesvirus biology. Trends Microbiol. 2007, 15, 426–433. [Google Scholar] [CrossRef]
- Atkinson, S.J.; Doberstein, S.K.; Pollard, T.D. Moving off the beaten track. Curr. Biol. 1992, 2, 326–328. [Google Scholar] [CrossRef]
- Kelleher, J.F.; Titus, M.A. Intracellular motility: How can we all work together? Curr. Biol. 1998, 8, R394–R397. [Google Scholar] [CrossRef]
- Langford, G.M. Actin- and microtubule-dependent organelle motors: Interrelationships between the two motility systems. Curr. Opin. Cell. Biol. 1995, 7, 82–88. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Actin dynamics. J. Cell. Sci. 2001, 114, 3–4. [Google Scholar]
- Pfaendtner, J.; Lyman, E.; Pollard, T.D.; Voth, G.A. Structure and dynamics of the actin filament. J. Mol. Biol. 2010, 396, 252–263. [Google Scholar]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and heads) of functional diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef]
- Lewis, T.L.; Mao, T.; Arnold, D.B. A role for myosin vi in the localization of axonal proteins. PLoS Biology 2011, 9, e1001021. [Google Scholar]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron. 2010, 68, 610–638. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu. Rev. Cell. Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef]
- Lüders, J.; Stearns, T. Microtubule-organizing centres: A re-evaluation. Nat. Rev. Mol. Cell. Biol. 2007, 8, 161–167. [Google Scholar] [CrossRef]
- Kirschner, M.W.; Mitchison, T. Microtubule dynamics. Nature 1986, 324, 621. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef]
- Lawrence, C.J.; Dawe, R.K.; Christie, K.R.; Cleveland, D.W.; Dawson, S.C.; Endow, S.A.; Goldstein, L.S.B.; Goodson, H.V.; Hirokawa, N.; Howard, J.; et al. A standardized kinesin nomenclature. J. Cell. Biol. 2004, 167, 19–22. [Google Scholar] [CrossRef]
- Petrásek, J.; Schwarzerová, K. Actin and microtubule cytoskeleton interactions. Curr. Opin. Plant. Biol. 2009, 12, 728–734. [Google Scholar] [CrossRef]
- Arnold, D.B. Actin and microtubule-based cytoskeletal cues direct polarized targeting of proteins in neurons. Sci. Signal. 2009, 2, pe49. [Google Scholar]
- Song, A.H.; Wang, D.; Chen, G.; Li, Y.; Luo, J.; Duan, S.; Poo, M.M. A selective filter for cytoplasmic transport at the axon initial segment. Cell 2009, 136, 1148–1160. [Google Scholar] [CrossRef]
- Leterrier, C.; Vacher, H.; Fache, M.P.; d'Ortoli, S.A.; Castets, F.; Autillo-Touati, A.; Dargent, B. End-binding proteins eb3 and eb1 link microtubules to ankyrin g in the axon initial segment. Proc. Natl. Acad. Sci. USA 2011, 108, 8826–8831. [Google Scholar]
- Nakata, T.; Hirokawa, N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell. Biol. 2003, 162, 1045–1055. [Google Scholar] [CrossRef]
- Hedstrom, K.L.; Ogawa, Y.; Rasband, M.N. Ankyring is required for maintenance of the axon initial segment and neuronal polarity. J. Cell. Biol. 2008, 183, 635–640. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell. Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell. Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef]
- De Regge, N.; Nauwynck, H.J.; Geenen, K.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Mettenleiter, T.; Favoreel, H.W. Alpha-herpesvirus glycoprotein d interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J. Cell. Biol. 2006, 174, 267–275. [Google Scholar]
- Kristensson, K.; Lycke, E.; Röyttä, M.; Svennerholm, B.; Vahlne, A. Neuritic transport of herpes simplex virus in rat sensory neurons in vitro. Effects of substances interacting with microtubular function and axonal flow [nocodazole, taxol and erythro-9–3-(2-hydroxynonyl)adenine]. J. Gen. Virol. 1986, 67, 2023–2028. [Google Scholar] [CrossRef]
- Liu, W.W.; Goodhouse, J.; Jeon, N.L.; Enquist, L.W. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS ONE 2008, 3, e2382. [Google Scholar] [CrossRef]
- Smith, G.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 2004, 101, 16034–16039. [Google Scholar] [CrossRef]
- Kardon, J.R.; Vale, R.D. Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell. Biol. 2009, 10, 854–865. [Google Scholar] [CrossRef]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes simplex virus type 1 capsid protein vp26 interacts with dynein light chains rp3 and tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar]
- Roberts, A.P.E.; Abaitua, F.; O'Hare, P.; McNab, D.; Rixon, F.J.; Pasdeloup, D. Differing roles of inner tegument proteins pul36 and pul37 during entry of herpes simplex virus type 1. J. Virol. 2009, 83, 105–116. [Google Scholar] [CrossRef]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.-H.; Bauerfeind, R.; Sodeik, B. The c terminus of the large tegument protein pul36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef]
- Shanda, S.K.; Wilson, D.W. Ul36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 2008, 82, 7388–7394. [Google Scholar]
- Abaitua, F.; Daikoku, T.; Crump, C.; Bolstad, M.; O'Hare, P. A single mutation responsible for temperature-sensitive entry and assembly defects in the vp1–2 protein of herpes simplex virus. J. Virol. 2011, 85, 2024. [Google Scholar] [CrossRef]
- Abaitua, F.; Souto, R.N.; Browne, H.; Daikoku, T.; O'Hare, P. Characterization of the herpes simplex virus (hsv)-1 tegument protein vp1–2 during infection with the hsv temperature-sensitive mutant tsb7. J. Gen. Virol. 2009, 90, 2353–2363. [Google Scholar] [CrossRef]
- Antinone, S.E.; Shubeita, G.T.; Coller, K.E.; Lee, J.I.; Haverlock-Moyns, S.; Gross, S.P.; Smith, G. The herpesvirus capsid surface protein, vp26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J. Virol. 2006, 80, 5494–5498. [Google Scholar]
- Desai, P.; DeLuca, N.A.; Person, S. Herpes simplex virus type 1 vp26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 1998, 247, 115–124. [Google Scholar] [CrossRef]
- Döhner, K.; Radtke, K.; Schmidt, S.; Sodeik, B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein vp26. J. Virol. 2006, 80, 8211–8224. [Google Scholar] [CrossRef]
- Desai, P.J. A null mutation in the ul36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 2000, 74, 11608–11618. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Mettenleiter, T. Essential function of the pseudorabies virus ul36 gene product is independent of its interaction with the ul37 protein. J. Virol. 2004, 78, 11879–11889. [Google Scholar]
- Vittone, V.; Diefenbach, E.; Triffett, D.; Douglas, M.W.; Cunningham, A.L.; Diefenbach, R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 2005, 79, 9566–9571. [Google Scholar] [CrossRef]
- Lee, J.H.; Vittone, V.; Diefenbach, E.; Cunningham, A.L.; Diefenbach, R.J. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 2008, 378, 347–354. [Google Scholar] [CrossRef]
- Loomis, J.S.; Bowzard, J.B.; Courtney, R.J.; Wills, J.W. Intracellular trafficking of the ul11 tegument protein of herpes simplex virus type 1. J. Virol. 2001, 75, 12209–12219. [Google Scholar] [CrossRef]
- Wagenaar, F.; Pol, J.M.; Peeters, B.; Gielkens, A.L.; de Wind, N.; Kimman, T.G. The us3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 1995, 76, 1851–1859. [Google Scholar] [CrossRef]
- Klupp, B.G.; Böttcher, S.; Granzow, H.; Kopp, M.; Mettenleiter, T. Complex formation between the ul16 and ul21 tegument proteins of pseudorabies virus. J. Virol. 2005, 79, 1510–1522. [Google Scholar]
- Kramer, T.; Greco, T.M.; Enquist, L.W.; Cristea, I.M. Proteomic characterization of pseudorabies virus extracellular virions. J. Virol. 2011, 85, 6427–6441. [Google Scholar] [CrossRef]
- Coller, K.E.; Lee, J.I.-H.; Ueda, A.; Smith, G. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the ul25 and vp1/2 proteins. J. Virol. 2007, 81, 11790–11797. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Miranda-Saksena, M.; Diefenbach, E.; Holland, D.J.; Boadle, R.A.; Armati, P.J.; Cunningham, A.L. Herpes simplex virus tegument protein us11 interacts with conventional kinesin heavy chain. J. Virol. 2002, 76, 3282–3291. [Google Scholar]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 us11 gene product with the cellular kinesin light-chain-related protein pat1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef]
- Lee, G.E.; Murray, J.W.; Wolkoff, A.W.; Wilson, D.W. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 2006, 80, 4264–4275. [Google Scholar] [CrossRef]
- Fuchs, W.; Granzow, H.; Klupp, B.G.; Kopp, M.; Mettenleiter, T. The ul48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 2002, 76, 6729–6742. [Google Scholar]
- Wolfstein, A.; Nagel, C.-H.; Radtke, K.; Döhner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef]
- Desai, P.; Sexton, G.L.; Huang, E.; Person, S. Localization of herpes simplex virus type 1 ul37 in the golgi complex requires ul36 but not capsid structures. J. Virol. 2008, 82, 11354–11361. [Google Scholar] [CrossRef]
- Szilágyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72, 661–668. [Google Scholar] [CrossRef]
- Babic, N.; Klupp, B.; Brack, A.; Mettenleiter, T.; Ugolini, G.; Flamand, A. Deletion of glycoprotein ge reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology 1996, 219, 279–284. [Google Scholar] [CrossRef]
- Brideau, A.D.; Card, J.P.; Enquist, L.W. Role of pseudorabies virus us9, a type ii membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 2000, 74, 834–845. [Google Scholar] [CrossRef]
- Card, J.P.; Levitt, P.; Enquist, L.W. Different patterns of neuronal infection after intracerebral injection of two strains of pseudorabies virus. J. Virol. 1998, 72, 4434–4441. [Google Scholar]
- Kritas, S.K.; Nauwynck, H.J.; Pensaert, M.B. Dissemination of wild-type and gc-, ge-and gi-deleted mutants of aujeszky's disease virus in the maxillary nerve and trigeminal ganglion of pigs after intranasal inoculation. J. Gen. Virol. 1995, 76, 2063–2066. [Google Scholar] [CrossRef]
- Mulder, W.; Pol, J.; Kimman, T.; Kok, G.; Priem, J.; Peeters, B. Glycoprotein d-negative pseudorabies virus can spread transneuronally via direct neuron-to-neuron transmission in its natural host, the pig, but not after additional inactivation of ge or gi. J. Virol. 1996, 70, 2191–2200. [Google Scholar]
- Mulder, W.A.; Jacobs, L.; Priem, J.; Kok, G.L.; Wagenaar, F.; Kimman, T.G.; Pol, J.M. Glycoprotein ge-negative pseudorabies virus has a reduced capability to infect second- and third-order neurons of the olfactory and trigeminal routes in the porcine central nervous system. J. Gen. Virol. 1994, 75, 3095–3106. [Google Scholar] [CrossRef]
- Whealy, M.E.; Card, J.P.; Robbins, A.K.; Dubin, J.R.; Rziha, H.J.; Enquist, L.W. Specific pseudorabies virus infection of the rat visual system requires both gi and gp63 glycoproteins. J. Virol. 1993, 67, 3786–3797. [Google Scholar]
- Kritas, S.K.; Pensaert, M.B.; Mettenleiter, T. Role of envelope glycoproteins gi, gp63 and giii in the invasion and spread of aujeszky's disease virus in the olfactory nervous pathway of the pig. J. Gen. Virol. 1994, 75, 2319–2327. [Google Scholar] [CrossRef]
- Ch'ng, T.H.; Enquist, L.W. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 2005, 79, 10875–10889. [Google Scholar] [CrossRef]
- Tirabassi, R.S.; Enquist, L.W. Role of envelope protein ge endocytosis in the pseudorabies virus life cycle. J. Virol. 1998, 72, 4571–4579. [Google Scholar]
- Tirabassi, R.S.; Townley, R.A.; Eldridge, M.G.; Enquist, L.W. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of ge: Evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 1997, 71, 6455–6464. [Google Scholar]
- Brideau, A.D.; Enquist, L.; Tirabassi, R.S. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 2000, 17, 69–82. [Google Scholar] [CrossRef]
- Tirabassi, R.S.; Enquist, L.W. Mutation of the yxxl endocytosis motif in the cytoplasmic tail of pseudorabies virus ge. J. Virol. 1999, 73, 2717–2728. [Google Scholar]
- Brideau, A.D.; Banfield, B.W.; Enquist, L.W. The us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type ii membrane protein. J. Virol. 1998, 72, 4560–4570. [Google Scholar]
- Lyman, M.G.; Curanovic, D.; Enquist, L.W. Targeting of pseudorabies virus structural proteins to axons requires association of the viral us9 protein with lipid rafts. PLoS Pathog. 2008, 4, e1000065. [Google Scholar] [CrossRef]
- Tomishima, M.J.; Smith, G.; Enquist, L.W. Sorting and transport of alpha herpesviruses in axons. Traffic 2001, 2, 429–436. [Google Scholar] [CrossRef]
- Aizawa, H.; Sekine, Y.; Takemura, R.; Zhang, Z.; Nangaku, M.; Hirokawa, N. Kinesin family in murine central nervous system. J. Cell. Biol. 1992, 119, 1287–1296. [Google Scholar] [CrossRef]
- Okada, Y.; Yamazaki, H.; Sekine-Aizawa, Y.; Hirokawa, N. The neuron-specific kinesin superfamily protein kif1a is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 1995, 81, 769–780. [Google Scholar] [CrossRef]
- Lo, K.Y.; Kuzmin, A.; Unger, S.M.; Petersen, J.D.; Silverman, M.A. Kif1a is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 2011, 491, 168–173. [Google Scholar] [CrossRef]
- Pack-Chung, E.; Kurshan, P.T.; Dickman, D.K.; Schwarz, T.L. A drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat. Neurosci. 2007, 10, 980–989. [Google Scholar] [CrossRef]
- Yonekawa, Y.; Harada, A.; Okada, Y.; Funakoshi, T.; Kanai, Y.; Takei, Y.; Terada, S.; Noda, T.; Hirokawa, N. Defect in synaptic vesicle precursor transport and neuronal cell death in kif1a motor protein-deficient mice. J. Cell. Biol. 1998, 141, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.H.; Hedgecock, E.M. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in c. Elegans. Cell 1991, 65, 837–847. [Google Scholar] [CrossRef]
- Klopfenstein, D.R.; Tomishige, M.; Stuurman, N.; Vale, R.D. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the unc104 kinesin motor. Cell 2002, 109, 347–358. [Google Scholar]
- Smith, G.; Gross, S.P.; Enquist, L.W. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 3466–3470. [Google Scholar] [CrossRef]
- Tannous, R.; Grose, C. Calculation of the anterograde velocity of varicella-zoster virions in a human sciatic nerve during shingles. J. Infect. Dis. 2011, 203, 324–326. [Google Scholar] [CrossRef]
- Roberts, K.L.; Baines, J.D. Myosin va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J. Virol. 2010, 84, 9889–9896. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell. Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the us3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef]
- Liu, M.; Schmidt, E.E.; Halford, W.P. Icp0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS ONE 2010, 5, e10975. [Google Scholar]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The mechanism of ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Grubb, M.S.; Burrone, J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 2010, 465, 1070–1074. [Google Scholar]
- Kuba, H.; Oichi, Y.; Ohmori, H. Presynaptic activity regulates na(+) channel distribution at the axon initial segment. Nature 2010, 465, 1075–1078. [Google Scholar] [CrossRef]
- Wimmer, V.C.; Reid, C.A.; So, E.Y.-W.; Berkovic, S.F.; Petrou, S. Axon initial segment dysfunction in epilepsy. J. Physiol. 2010, 588, 1829–1840. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 2, 77–93. [Google Scholar]
- McCarthy, K.M.; Tank, D.W.; Enquist, L.W. Pseudorabies virus infection alters neuronal activity and connectivity in vitro. PLoS Patho. 2009, 5, e1000640. [Google Scholar] [CrossRef]
- Kramer, T.; Enquist, L.W. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe. 2012, 11, 504–514. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the rig-i-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kramer, T.; Enquist, L.W. Directional Spread of Alphaherpesviruses in the Nervous System. Viruses 2013, 5, 678-707. https://doi.org/10.3390/v5020678
Kramer T, Enquist LW. Directional Spread of Alphaherpesviruses in the Nervous System. Viruses. 2013; 5(2):678-707. https://doi.org/10.3390/v5020678
Chicago/Turabian StyleKramer, Tal, and Lynn W. Enquist. 2013. "Directional Spread of Alphaherpesviruses in the Nervous System" Viruses 5, no. 2: 678-707. https://doi.org/10.3390/v5020678
APA StyleKramer, T., & Enquist, L. W. (2013). Directional Spread of Alphaherpesviruses in the Nervous System. Viruses, 5(2), 678-707. https://doi.org/10.3390/v5020678