Structure, Function and Dynamics in Adenovirus Maturation


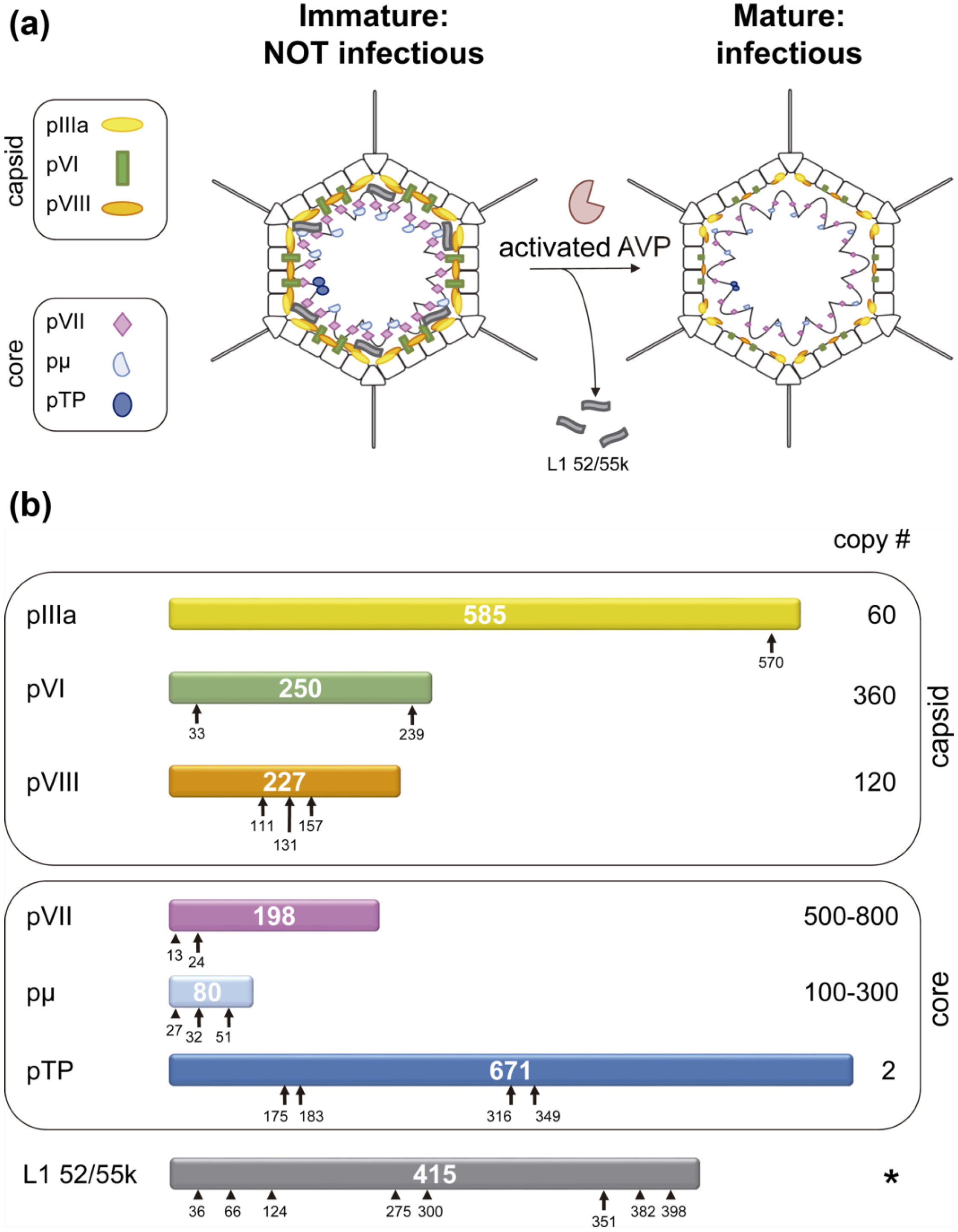{kind=link}
{kind=link}
{kind=link}
{kind=link}
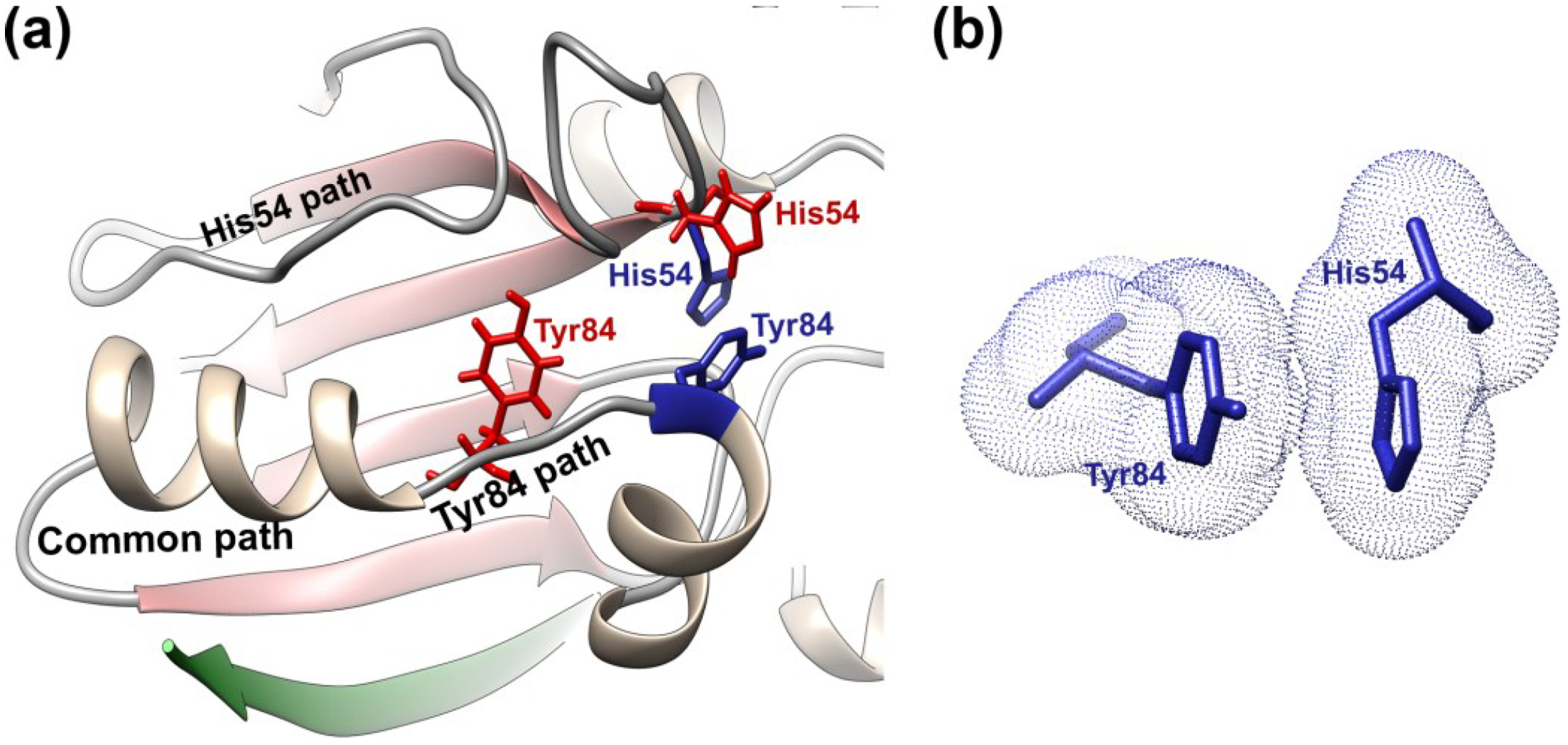{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Adenovirus
2. Players in Adenovirus Maturation: The Protease and Its Substrates

3. Unveiling the Enzymatic Mechanism of AVP


3.1. Discovery of the AVP Cofactors
3.2. Structure of AVP

3.3. Interactions of AVP with Its Cofactors
3.4. AVP Activation Pathways

3.5. AVP Activation in Its Biological Context
3.6. AVP Function in Its Biological Context

3.7. Using AVP as a Target for Anti-Adenovirus Drugs
4. Effect of Adenovirus Maturation on the Viral Particle
4.1. Structural Changes Induced by Maturation of the Viral Particle

4.2. Stability Changes Induced by Maturation of the Viral Particle
4.3. Release of Packaging Scaffold Protein L1 52/55k
5. Proteolytic Processing of Pre-Terminal Protein
6. Concluding Remarks and Remaining Questions
6.1. Enzymology and Mode of Action of AVP
6.2. Role of Maturation in the AdV Infectious Cycle
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Berk, A.J. Adenoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2355–2394. [Google Scholar]
- Harrach, B.; Benkő, M.; Both, G.; Brown, M.; Davison, A.; Echavarría, M.; Hess, M.; Jones, M.; Kajon, A.; Lehmkuhl, H.; et al. Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; King, A., Adams, M., Carstens, E., Lefkowitz, E., Eds.; Elsevier: San Diego, CA, USA, 2011; pp. 95–111. [Google Scholar]
- Lasaro, M.O.; Ertl, H.C. New insights on adenovirus as vaccine vectors. Mol. Ther. 2009, 17, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.A.F.V.; de Vries, A.A.F. Adenovirus: From foe to friend. Rev. Med. Virol. 2006, 16, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Curiel, D.T. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Castón, J.R.; Carrascosa, J.L. The basic architecture of viruses. Sub-Cell. Biochem. 2013, 68, 53–75. [Google Scholar]
- San Martín, C. Latest Insights on Adenovirus Structure and Assembly. Viruses 2012, 4, 847–877. [Google Scholar]
- Liu, H.; Jin, L.; Koh, S.B.; Atanasov, I.; Schein, S.; Wu, L.; Zhou, Z.H. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 2010, 329, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Natchiar, S.K.; Stewart, P.L.; Nemerow, G.R. Crystal structure of human adenovirus at 3.5 A resolution. Science 2010, 329, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Nemerow, G.R. Structures and organization of adenovirus cement proteins provide insights into the role of capsid maturation in virus entry and infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K. New structural model of adenoviral cement proteins is not yet concrete. Proc. Natl. Acad. Sci. USA 2014, 111, E4542–E4543. [Google Scholar] [CrossRef] [PubMed]
- Saban, S.D.; Silvestry, M.; Nemerow, G.R.; Stewart, P.L. Visualization of α-helices in a 6 Å resolution cryoEM structure of adenovirus allows refinement of capsid protein assignments. J. Virol. 2006, 80, 12049–12059. [Google Scholar] [CrossRef] [PubMed]
- San Martín, C.; Glasgow, J.N.; Borovjagin, A.; Beatty, M.S.; Kashentseva, E.A.; Curiel, D.T.; Marabini, R.; Dmitriev, I.P. Localization of the N-terminus of minor coat protein IIIa in the adenovirus capsid. J. Mol. Biol. 2008, 383, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H.W.; Marabini, R.; Lanzavecchia, S.; Cantele, F.; Rutten, T.; Fuller, S.D.; Carazo, J.M.; Burnett, R.M.; San Martín, C. Classification of single-projection reconstructions for cryo-electron microscopy data of icosahedral viruses. J. Struct. Biol. 2005, 151, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.C.; Hearing, P. Adenovirus structural protein IIIa is involved in the serotype specificity of viral DNA packaging. J. Virol. 2011, 85, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; Graziano, V.; Pérez-Berná, A.J.; McGrath, W.J.; Flint, S.J.; San Martín, C.; Xie, X.S.; Mangel, W.F. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space: IV. Viral proteinase slides along DNA to locate and process its substrates. J. Biol. Chem. 2013, 288, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oostrum, J.; Burnett, R.M. Molecular composition of the adenovirus type 2 virion. J. Virol. 1985, 56, 439–448. [Google Scholar] [PubMed]
- Benevento, M.; di Palma, S.; Snijder, J.; Moyer, C.L.; Reddy, V.S.; Nemerow, G.R.; Heck, A.J. Adenovirus composition, proteolysis, and disassembly studied by in-depth qualitative and quantitative proteomics. J. Biol. Chem. 2014, 289, 11421–11430. [Google Scholar] [CrossRef] [PubMed]
- Vayda, M.E.; Rogers, A.E.; Flint, S.J. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res. 1983, 11, 441–460. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Weber, J. Structure of adenovirus chromatin. Biochim. Biophys. Acta 1982, 696, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N. Adenovirus receptors: Implications for targeting of viral vectors. Trends Pharmacol. Sci. 2012, 33, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.Y.; Boucke, K.; Suomalainen, M.; Stidwill, R.P.; Greber, U.F. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J. Virol. 2000, 74, 7085–7095. [Google Scholar] [CrossRef] [PubMed]
- Lindert, S.; Silvestry, M.; Mullen, T.M.; Nemerow, G.R.; Stewart, P.L. Cryo-electron microscopy structure of an adenovirus-integrin complex indicates conformational changes in both penton base and integrin. J. Virol. 2009, 83, 11491–11501. [Google Scholar] [CrossRef] [PubMed]
- Puntener, D.; Engelke, M.F.; Ruzsics, Z.; Strunze, S.; Wilhelm, C.; Greber, U.F. Stepwise loss of fluorescent core protein V from human adenovirus during entry into cells. J. Virol. 2011, 85, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog. 2010, 6, e1000808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burckhardt, C.J.; Suomalainen, M.; Schoenenberger, P.; Boucke, K.; Hemmi, S.; Greber, U.F. Drifting motions of the adenovirus receptor CAR and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 2011, 10, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, O.; Galan, D.L.; Wodrich, H.; Wiethoff, C.M. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 2010, 402, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, M.; Luisoni, S.; Boucke, K.; Bianchi, S.; Engel, D.A.; Greber, U.F. A direct and versatile assay measuring membrane penetration of adenovirus in single cells. J. Virol. 2013, 87, 12367–12379. [Google Scholar] [CrossRef] [PubMed]
- Bremner, K.H.; Scherer, J.; Yi, J.; Vershinin, M.; Gross, S.P.; Vallee, R.B. Adenovirus transport via direct interaction of cytoplasmic dynein with the viral capsid hexon subunit. Cell Host Microbe 2009, 6, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Suomalainen, M.; Stidwill, R.P.; Boucke, K.; Ebersold, M.W.; Helenius, A. The role of the nuclear pore complex in adenovirus DNA entry. EMBO J. 1997, 16, 5998–6007. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Mosberger, N.; Fornerod, M.; Stidwill, R.P.; Greber, U.F. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 2001, 3, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, M.; Nakano, M.Y.; Keller, S.; Boucke, K.; Stidwill, R.P.; Greber, U.F. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 1999, 144, 657–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Walkiewicz, M.P.; Morral, N.; Engel, D.A. Accurate single-day titration of adenovirus vectors based on equivalence of protein VII nuclear dots and infectious particles. J. Virol. Methods 2009, 159, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Gustin, K.E.; Imperiale, M.J. Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J. Virol. 1998, 72, 7860–7870. [Google Scholar] [PubMed]
- Guimet, D.; Hearing, P. The Adenovirus L4-22K Protein Has Distinct Functions in the Posttranscriptional Regulation of Gene Expression and Encapsidation of the Viral Genome. J. Virol. 2013, 87, 7688–7699. [Google Scholar] [CrossRef]
- Wu, K.; Guimet, D.; Hearing, P. The Adenovirus L4-33K Protein Regulates both Late Gene Expression Patterns and Viral DNA Packaging. J. Virol. 2013, 87, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- Ostapchuk, P.; Almond, M.; Hearing, P. Characterization of Empty adenovirus particles assembled in the absence of a functional adenovirus IVa2 protein. J. Virol. 2011, 85, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Imperiale, M.J. Interaction of the adenovirus IVa2 protein with viral packaging sequences. J. Virol. 2000, 74, 2687–2693. [Google Scholar] [CrossRef] [PubMed]
- Ostapchuk, P.; Yang, J.; Auffarth, E.; Hearing, P. Functional interaction of the adenovirus IVa2 protein with adenovirus type 5 packaging sequences. J. Virol. 2005, 79, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Perez-Romero, P.; Tyler, R.E.; Abend, J.R.; Dus, M.; Imperiale, M.J. Analysis of the interaction of the adenovirus L1 52/55-kilodalton and IVa2 proteins with the packaging sequence in vivo and in vitro. J. Virol. 2005, 79, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Ostapchuk, P.; Anderson, M.E.; Chandrasekhar, S.; Hearing, P. The L4 22-kilodalton protein plays a role in packaging of the adenovirus genome. J. Virol. 2006, 80, 6973–6981. [Google Scholar] [CrossRef] [PubMed]
- Hasson, T.B.; Ornelles, D.A.; Shenk, T. Adenovirus L1 52- and 55-kilodalton proteins are present within assembling virions and colocalize with nuclear structures distinct from replication centers. J. Virol. 1992, 66, 6133–6142. [Google Scholar] [PubMed]
- Ishibashi, M.; Maizel, J.V., Jr. The polypeptides of adenovirus. V. Young virions, structural intermediate between top components and aged virions. Virology 1974, 57, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.M. Role of endoprotease in adenovirus infection. In Adenoviruses: Basic Biology to Gene Therapy; Seth, P., Ed.; R.G. Landes: Austin, TX, USA, 1999; pp. 79–83. [Google Scholar]
- Cotten, M.; Weber, J.M. The adenovirus protease is required for virus entry into host cells. Virology 1995, 213, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, M.; Imelli, N.; Boucke, K.; Amstutz, B.; Meier, O.; Greber, U.F. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 2008, 9, 2265–2278. [Google Scholar] [CrossRef] [PubMed]
- Weber, J. Genetic analysis of adenovirus type 2 III. Temperature sensitivity of processing viral proteins. J. Virol. 1976, 17, 462–471. [Google Scholar] [PubMed]
- Mangel, W.F.; Toledo, D.L.; Brown, M.T.; Martin, J.H.; McGrath, W.J. Characterization of three components of human adenovirus proteinase activity in vitro. J. Biol. Chem. 1996, 271, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Blanche, F.; Monegier, B.; Faucher, D.; Duchesne, M.; Audhuy, F.; Barbot, A.; Bouvier, S.; Daude, G.; Dubois, H.; Guillemin, T.; et al. Polypeptide composition of an adenovirus type 5 used in cancer gene therapy. J. Chromatogr. A 2001, 921, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Challberg, M.D.; Kelly, T.J., Jr. Processing of the adenovirus terminal protein. J. Virol. 1981, 38, 272–277. [Google Scholar] [PubMed]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.; Kemp, G. The active adenovirus protease is the intact L3 23K protein. J. Gen. Virol. 1993, 74, 1415–1420. [Google Scholar] [CrossRef]
- Anderson, C.W. The proteinase polypeptide of adenovirus serotype 2 virions. Virology 1990, 177, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.T.; McGrath, W.J.; Toledo, D.L.; Mangel, W.F. Different modes of inhibition of human adenovirus proteinase, probably a cysteine proteinase, by bovine pancreatic trypsin inhibitor. FEBS Lett. 1996, 388, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Arcos, R. Interaction of the adenovirus major core protein precursor, pVII, with the viral DNA packaging machinery. Virology 2005, 334, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Berná, A.J.; Mangel, W.F.; McGrath, W.J.; Graziano, V.; Flint, J.; San Martín, C. Processing of the L1 52/55k protein by the adenovirus protease: A new substrate and new insights into virion maturation. J. Virol. 2014, 88, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Yeh-Kai, L.; Akusjarvi, G.; Alestrom, P.; Pettersson, U.; Tremblay, M.; Weber, J. Genetic identification of an endopeptidase encoded by the adenovirus genome. J. Mol. Biol. 1983, 167, 217–222. [Google Scholar] [CrossRef]
- Anderson, C.W. Expression and purification of the adenovirus proteinase polypeptide and of a synthetic proteinase substrate. Protein Express. Purif. 1993, 4, 8–15. [Google Scholar] [CrossRef]
- Tihanyi, K.; Bourbonniere, M.; Houde, A.; Rancourt, C.; Weber, J.M. Isolation and properties of adenovirus type 2 proteinase. J. Biol. Chem. 1993, 268, 1780–1785. [Google Scholar] [PubMed]
- Webster, A.; Hay, R.T.; Kemp, G. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 1993, 72, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, C.; Keyvani-Amineh, H.; Sircar, S.; Labrecque, P.; Weber, J.M. Proline 137 is critical for adenovirus protease encapsidation and activation but not enzyme activity. Virology 1995, 209, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Imelli, N.; Ruzsics, Z.; Puntener, D.; Gastaldelli, M.; Greber, U.F. Genetic reconstitution of the human adenovirus type 2 temperature-sensitive 1 mutant defective in endosomal escape. Virol. J. 2009, 6, 174. [Google Scholar] [CrossRef] [PubMed]
- Diouri, M.; Keyvani-Amineh, H.; Geoghegan, K.F.; Weber, J.M. Cleavage efficiency by adenovirus protease is site-dependent. J. Biol. Chem. 1996, 271, 32511–32514. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.; Russell, S.; Talbot, P.; Russell, W.C.; Kemp, G.D. Characterization of the adenovirus proteinase: Substrate specificity. J. Gen. Virol. 1989, 70, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.; Leith, I.R.; Nicholson, J.; Hounsell, J.; Hay, R.T. Role of preterminal protein processing in adenovirus replication. J. Virol. 1997, 71, 6381–6389. [Google Scholar] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Leytus, S.P.; Melhado, L.L.; Mangel, W.F. Rhodamine-based compounds as fluorogenic substrates for serine proteases. Biochem. J. 1983, 209, 299–307. [Google Scholar] [PubMed]
- Leytus, S.P.; Patterson, W.L.; Mangel, W.F. New class of sensitive, specific, and selective substrates for serine proteinases: Fluorogenic, amino acid peptide derivatives of Rhodamine. Biochem. J. 1983, 215, 253–260. [Google Scholar] [PubMed]
- Webster, A.; Russell, W.C.; Kemp, G.D. Characterization of the adenovirus proteinase; development and use of a specific peptide assay. J. Gen. Virol. 1989, 70, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Mangel, W.F.; McGrath, W.J.; Toledo, D.L.; Anderson, C.W. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 1993, 361, 274–275. [Google Scholar] [CrossRef] [PubMed]
- McGrath, W.J.; Abola, A.P.; Toledo, D.L.; Brown, M.T.; Mangel, W.F. Characterization of human adenovirus proteinase activity in disrupted virus particles. Virology 1996, 217, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bajpayee, N.S.; McGrath, W.J.; Mangel, W.F. Interaction of the adenovirus proteinase with protein cofactors with high negative charge densities. Biochemistry 2005, 44, 8721–8729. [Google Scholar] [CrossRef] [PubMed]
- McGrath, W.J.; Baniecki, M.L.; Li, C.; McWhirter, S.M.; Brown, M.T.; Toledo, D.L.; Mangel, W.F. Human adenovirus proteinase: DNA binding and stimulation of proteinase activity by DNA. Biochemistry 2001, 40, 13237–13245. [Google Scholar] [CrossRef] [PubMed]
- Joshua-Tor, L.; Xu, H.E.; Johnston, S.A.; Rees, D.C. Crystal structure of a conserved protease that binds DNA: The bleomycin hydrolase, Gal6. Science 1995, 269, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Staufenbiel, M.; Epple, P.; Deppert, W. Progressive reorganization of the host cell cytoskeleton during adenovirus infection. J. Virol. 1986, 60, 1186–1191. [Google Scholar] [PubMed]
- Chen, P.H.; Ornelles, D.A.; Shenk, T. The adenovirus L3 23-Kilodalton proteinase cleaves the amino-terminal head domain from cytokeratin 18 and disrupts the cytokeratin network of HeLa cells. J. Virol. 1993, 67, 3507–3514. [Google Scholar] [PubMed]
- Brown, M.T.; McBride, K.M.; Baniecki, M.L.; Reich, N.C.; Marriott, G.; Mangel, W.F. Actin can act as a cofactor for a viral proteinase, in the cleavage of the cytoskeleton. J. Biol. Chem. 2002, 277, 46298–46303. [Google Scholar] [CrossRef] [PubMed]
- Schutt, C.E.; Myslik, J.C.; Rozycki, M.D.; Goonesekere, N.C.; Lindberg, U. The structure of crystalline profilin-beta-actin. Nature 1993, 365, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Akusjarvi, G.; Persson, H.J. Gene and mRNA for precursor polypeptide VI from adenovirus type 2. J. Virol. 1981, 38, 469–482. [Google Scholar] [PubMed]
- Cai, F.; Weber, J.M. Organization of the avian adenovirus genome and the structure of its endopeptidase. Virology 1993, 196, 358–362. [Google Scholar] [CrossRef]
- McGrath, W.J.; Ding, J.; Sweet, R.M.; Mangel, W.F. Preparation and crystallization of a complex between human adenovirus serotype 2 proteinase and its 11-amino-acid cofactor pVIc. J. Struct. Biol. 1996, 117, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; McGrath, W.J.; Sweet, R.M.; Mangel, W.F. Crystal structure of the human adenovirus proteinase with its 11 amino acid cofactor. EMBO J. 1996, 15, 1778–1783. [Google Scholar] [PubMed]
- McGrath, W.J.; Ding, J.; Sweet, R.M.; Mangel, W.F. Crystallographic structure at 1.6-Å resolution of the human adenovirus proteinase in a covalent complex with its 11-amino-acid peptide cofactor: Insights on a new fold. Biochem. Biophys. Acta 2003, 1648, 1–11. [Google Scholar] [PubMed]
- Mangel, W.F.; Toledo, D.L.; Ding, J.; Sweet, R.M.; McGrath, W.J. Temporal and spatial control of the adenovirus proteinase by both a peptide and the viral DNA. Trends Biochem. Sci. 1997, 22, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Storer, A.C.; Menard, R. Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 1994, 244, 486–500. [Google Scholar] [PubMed]
- Robertus, J.D.; Kraut, J.; Alden, R.A.; Birktoft, J.J. Subtilisin; a stereochemical mechanism involving transition-state stabilization. Biochemistry 1972, 11, 4293–4303. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.; Kalk, K.H.; Swen, H.M. Binding of chloromethyl ketone substrate analogues to crystalline papain. Biochemistry 1976, 15, 3731–3738. [Google Scholar] [CrossRef] [PubMed]
- Baniecki, M.L.; McGrath, W.J.; Mangel, W.F. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space: III. Atomic resolution structure of the nascent form of the adenovirus proteinase. J. Biol. Chem. 2013, 288, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mangel, W.F.; McGrath, W.J.; Perek, J.L.; Lee, D.W.; Takamoto, K.; Chance, M.R. DNA binding provides a molecular strap activating the adenovirus proteinase. Mol. Cell. Proteomics 2004, 3, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mangel, W.F.; Sullivan, M.; Takamoto, K.G.; Chance, M.R. Mapping a functional viral protein in solution using synchrotron X-ray footprinting technology. Synchrotron Radiat. News 2005, 18, 25–34. [Google Scholar] [CrossRef]
- Baniecki, M.L.; McGrath, W.J.; McWhirter, S.M.; Li, C.; Toledo, D.L.; Pellicena, P.; Barnard, D.L.; Thorn, K.S.; Mangel, W.F. Interaction of the human adenovirus proteinase with its 11-amino acid cofactor pVIc. Biochemistry 2001, 40, 12349–12356. [Google Scholar] [CrossRef] [PubMed]
- Baniecki, M.L.; McGrath, W.J.; McWhirter, S.M.; Li, C.; Toledo, D.L.; Pellicena, P.; Barnard, D.L.; Thorn, K.S.; Mangel, W.F. Interaction of the human adenovirus proteinase with its 11-amino acid cofactor pVIc. Biochemistry 2001, 41, 430. [Google Scholar] [CrossRef]
- McGrath, W.J.; Baniecki, M.L.; Peters, E.; Green, D.T.; Mangel, W.F. Roles of two conserved cysteine residues in the activation of human adenovirus proteinase. Biochemistry 2001, 40, 14468–14474. [Google Scholar] [CrossRef] [PubMed]
- McGrath, W.J.; Aherne, K.S.; Mangel, W.F. In the virion, the 11-amino-acid peptide cofactor pVIc is covalently linked to the adenovirus proteinase. Virology 2002, 296, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C.; Precious, B. Nucleic acid-binding properties of adenovirus structural polypeptides. J. Gen. Virol. 1982, 63, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Record, M.T., Jr.; Lohman, M.L.; de Haseth, P. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976, 107, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Graziano, V.; McGrath, W.J.; Suomalainen, M.; Greber, U.F.; Freimuth, P.; Blainey, P.C.; Luo, G.; Xie, X.S.; Mangel, W.F. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space: I. Binding to DNA and to hexon of the precursor to protein VI, pVI, of human adenovirus. J. Biol. Chem. 2013, 288, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S. Principles of virion structure, function and assembly. In Structural Biology of Viruses; Chiu, W., Burnett, R.M., Garcea, R.L., Eds.; Oxford University Press: Oxford, UK, 1997; pp. 3–37. [Google Scholar]
- Mangenot, S.; Keller, S.; Radler, J. Transport of nucleosome core particles in semidilute DNA solutions. Biophys. J. 2003, 85, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Graziano, V.; Luo, G.; Blainey, P.C.; Pérez-Berná, A.J.; McGrath, W.J.; Flint, S.J.; San Martín, C.; Xie, X.S.; Mangel, W.F. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space: II. Adenovirus proteinase is activated in an unusual one-dimensional biochemical reaction. J. Biol. Chem. 2013, 288, 2068–2080. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.; Leith, I.R.; Hay, R.T. Activation of adenovirus-coded protease and processing of preterminal protein. J. Virol. 1994, 68, 7292–7300. [Google Scholar] [PubMed]
- Chatterjee, P.K.; Vayda, M.E.; Flint, S.J. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J. Mol. Biol. 1986, 188, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F. Virus assembly and disassembly: The adenovirus cysteine protease as a trigger factor. Rev. Med. Virol. 1998, 8, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; Luo, G.; Kou, S.C.; Mangel, W.F.; Verdine, G.L.; Bagchi, B.; Xie, X.S. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009, 16, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.D.; Kim, H.W.; Vargosdo, A.J.; Jefferies, B.C.; Arrobio, J.O.; Rindge, B.; Parrott, R.H.; Chanock, R.M. Infections in 18,000 infants and children in a controlled study of respiratory tract disease. I. Adenovirus pathogenicity in relation to serologic type and illness syndrome. Am. J. Epidemiol. 1969, 90, 484–500. [Google Scholar] [PubMed]
- Mallet, R.; Riberre, M.; Bonnenfant, F.; Labrune, B.; Reyrole, L. Les pneumopathies graves a adeno-virus. Arch. Fr. Pediatr. 1966, 23, 1057–1073. [Google Scholar]
- Kohoo, S.H.; Bailey, A.S.; de Jong, J.C.; Mandal, B.K. Adenovirus infections in human immunodeficiency virus-positive patients: Clinical features and molecular epidemiology. J. Infect. Dis. 1995, 172, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Mogabgab, W.J. Mycoplasma pneumonia and adenovirus respiratory illnesses in military and university personnel. Am. Rev. Respir. Dis. 1968, 97, 345–358. [Google Scholar] [PubMed]
- Gray, C.G.; Callahan, J.D.; Hawksworth, A.W.; Fisher, C.A.; Gaydos, J.C. Respiratory diseases among U.S. military personnel: Countering emerging threats. Emerg. Infect. Dis. 1999, 5, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Dhurandhar, N.V.; Allison, D.B.; Bown, R.L.; Israel, B.A.; Albu, J.B.; Augustus, A.S. Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. Int. J. Obes. 2005, 29, 281–286. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Acute respiratory disease associated with adenovirus serotype 14—Four states, 2006–2007. MMWR 2007, 56, 1181–1184. [Google Scholar]
- Mangel, W.F.; Brown, M.T.; Baniecki, M.L.; Barnard, D.L.; McGrath, W.J. Prevention of viral drug resistance by novel combination therapy. Curr. Opin. Investig. Drugs 2001, 2, 613–616. [Google Scholar] [PubMed]
- Mangel, W.F.; McGrath, W.J.; Brown, M.T.; Baniecki, M.L.; Barnard, D.L.; Pang, Y.P. A new form of antiviral combination therapy predicted to prevent resistance from arising, and a model system to test it. Curr. Med. Chem. 2001, 8, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Mangel, W.F.; Toledo, D.L.; Brown, M.T.; Ding, J.; Sweet, R.M.; Barnard, D.L.; McGrath, W.J. Adenovirus proteinase-antiviral target for triple-combination therapy on a single enzyme: Potential inhibitor-binding sites. In Proteases as Targets for Therapy; von der Helm, K., Korant, B.D., Cheronis, J.C., Eds.; Springer: Berlin, Germany, 2000; Volume 140, pp. 145–158. [Google Scholar]
- Pang, Y.-P.; Xu, K.; Kollmeyer, T.M.; Perola, E.; McGrath, W.J.; Green, D.T.; Mangel, W.F. Discovery of a new inhibitor lead of adenovirus proteinase: Steps toward selective, irreversible inhibitors of cysteine proteinases. FEBS Lett. 2001, 502, 93–97. [Google Scholar] [CrossRef] [PubMed]
- McGrath, W.J.; Graziano, V.; Mangel, W.F. First generation inhibitors of the adenovirus proteinase. FEBS Lett. 2013, 587, 2332–2339. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Johnson, J.E. Virus Maturation. Annu. Rev. Biophys. 2012, 41, 473–496. [Google Scholar] [CrossRef]
- Johnson, J.E. Virus particle maturation: Insights into elegantly programmed nanomachines. Curr. Opin. Struct. Biol. 2010, 20, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Silvestry, M.; Lindert, S.; Smith, J.G.; Maier, O.; Wiethoff, C.M.; Nemerow, G.R.; Stewart, P.L. Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J. Virol. 2009, 83, 7375–7383. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Berná, A.J.; Marabini, R.; Scheres, S.H.W.; Menéndez-Conejero, R.; Dmitriev, I.P.; Curiel, D.T.; Mangel, W.F.; Flint, S.J.; San Martín, C. Structure and uncoating of immature adenovirus. J. Mol. Biol. 2009, 392, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Snijder, J.; Benevento, M.; Moyer, C.L.; Reddy, V.; Nemerow, G.R.; Heck, A.J. The cleaved N-terminus of pVI binds peripentonal hexons in mature adenovirus. J. Mol. Biol. 2014, 426, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Hannan, C.; Raptis, L.H.; Dery, C.V.; Weber, J. Biological and structural studies with an adenovirus type 2 temperature-sensitive mutant defective for uncoating. Intervirology 1983, 19, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Berná, A.J.; Ortega-Esteban, A.; Menéndez-Conejero, R.; Winkler, D.C.; Menéndez, M.; Steven, A.C.; Flint, S.J.; de Pablo, P.J.; San Martín, C. The role of capsid maturation on adenovirus priming for sequential uncoating. J. Biol. Chem. 2012, 287, 31582–31595. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Chiu, W. DNA packaging and delivery machines in tailed bacteriophages. Curr. Opin. Struct. Biol. 2007, 17, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M. Poliovirus cell entry: Common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Esteban, A.; Pérez-Berná, A.J.; Menéndez-Conejero, R.; Flint, S.J.; San Martín, C.; de Pablo, P.J. Monitoring dynamics of human adenovirus disassembly induced by mechanical fatigue. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.; Ocheretina, O.; Schoggins, J.W.; Falck-Pedersen, E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 2007, 81, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Gustin, K.E.; Lutz, P.; Imperiale, M.J. Interaction of the adenovirus L1 52/55-kilodalton protein with the IVa2 gene product during infection. J. Virol. 1996, 70, 6463–6467. [Google Scholar] [PubMed]
- Rekosh, D.M.; Russell, W.C.; Bellet, A.J.; Robinson, A.J. Identification of a protein linked to the ends of adenovirus DNA. Cell 1977, 11, 283–295. [Google Scholar] [CrossRef]
- Stillman, B.W.; Lewis, J.B.; Chow, L.T.; Mathews, M.B.; Smart, J.E. Identification of the gene and mRNA for the adenovirus terminal protein precursor. Cell 1981, 23, 497–508. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H.; Hay, R.T. Adenovirus DNA replication. In Adenoviruses: Model and Vectors in Virus Host Interactions. Current Topics in Microbiology and Immunology; Doerfler, W., Böhm, P., Eds.; Springer-Verlag: Heidelberg, Germany, 2003; Volume 272, pp. 131–164. [Google Scholar]
- Khittoo, G.; Delorme, L.; Dery, C.V.; Tremblay, M.L.; Weber, J.M.; Bibor-Hardy, V.; Simard, R. Role of the nuclear matrix in adenovirus maturation. Virus Res. 1986, 4, 391–403. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; van der Vliet, P.C. A precursor terminal protein-trinucleotide intermediate during initiation of adenovirus DNA replication: Regeneration of molecular ends in vitro by a jumping back mechanism. EMBO J. 1994, 13, 5786–5792. [Google Scholar]
- Pronk, R.; Stuiver, M.H.; van der Vliet, P.C. Adenovirus DNA replication: The function of the covalently bound terminal protein. Chromosoma 1992, 102, S39–S45. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.E.; Chahal, J.S.; Flint, S.J. Reduced infectivity of adenovirus type 5 particles and degradation of entering viral genomes associated with incomplete processing of the preterminal protein. J. Virol. 2012, 86, 13554–13565. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrandt, W. Cryo-EM enters a new era. eLife 2014, 3, e03678. [Google Scholar]
- Uetrecht, C.; Heck, A.J. Modern biomolecular mass spectrometry and its role in studying virus structure, dynamics, and assembly. Angew. Chem. Int. Ed. Engl. 2011, 50, 8248–8262. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangel, W.F.; San Martín, C. Structure, Function and Dynamics in Adenovirus Maturation. Viruses 2014, 6, 4536-4570. https://doi.org/10.3390/v6114536
Mangel WF, San Martín C. Structure, Function and Dynamics in Adenovirus Maturation. Viruses. 2014; 6(11):4536-4570. https://doi.org/10.3390/v6114536
Chicago/Turabian StyleMangel, Walter F., and Carmen San Martín. 2014. "Structure, Function and Dynamics in Adenovirus Maturation" Viruses 6, no. 11: 4536-4570. https://doi.org/10.3390/v6114536
APA StyleMangel, W. F., & San Martín, C. (2014). Structure, Function and Dynamics in Adenovirus Maturation. Viruses, 6(11), 4536-4570. https://doi.org/10.3390/v6114536