
The A, B, Cs of Herpesvirus Capsids


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Chemical Inhibition
2.3. Transmission Electron Microscopy
2.4. Virus and Capsid Purification
2.5. Cryo-Electron Microscopy
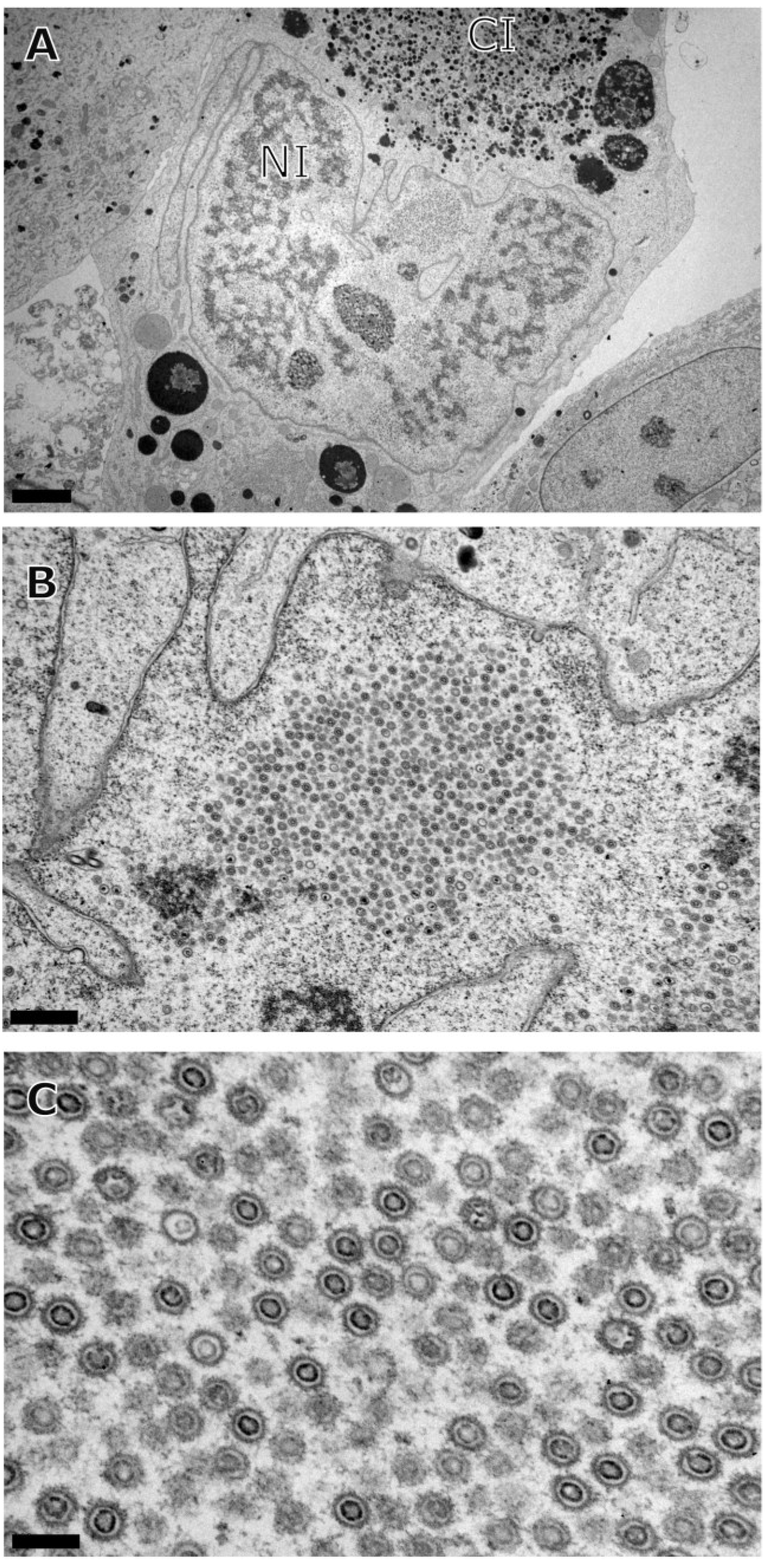
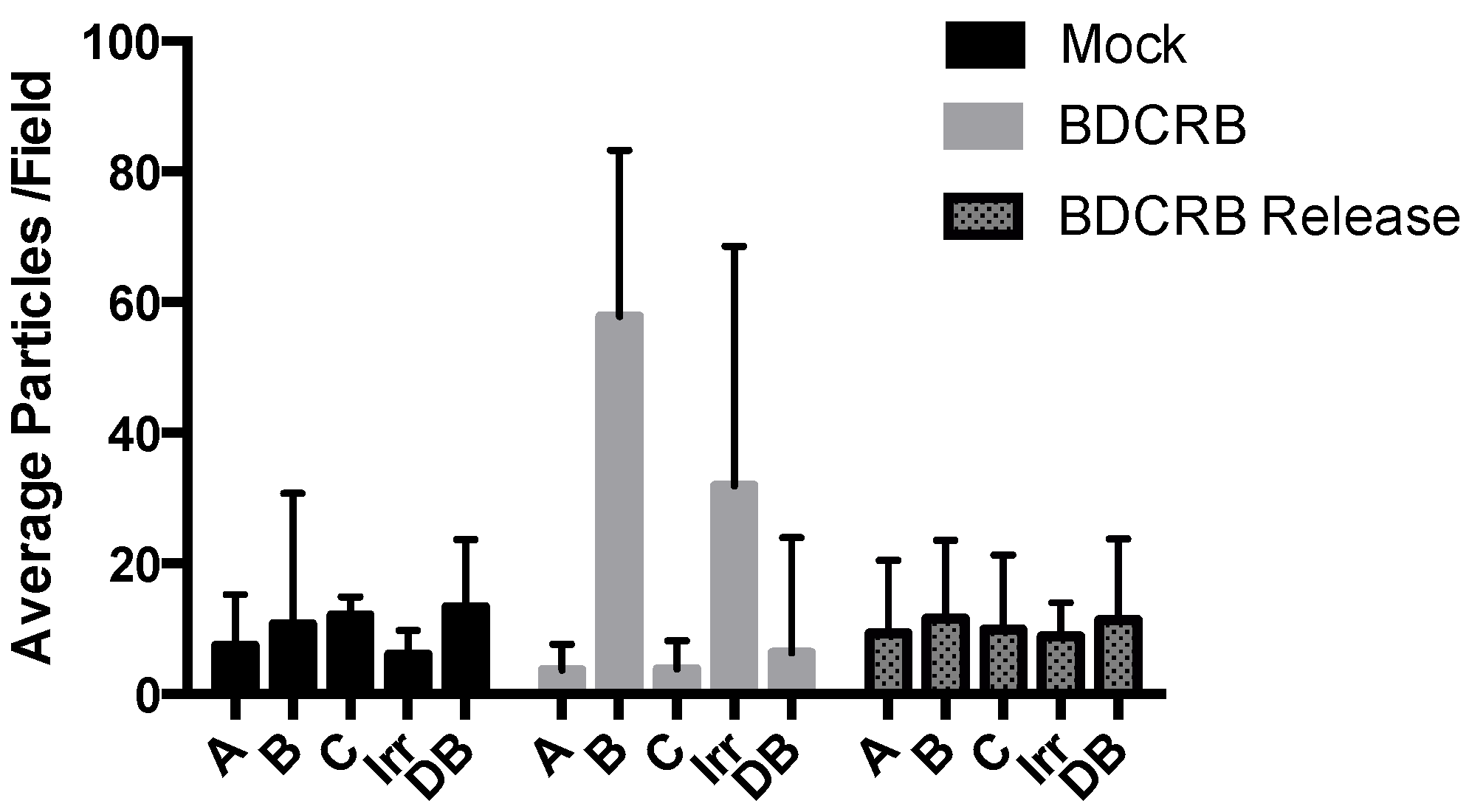
3. Results








{kind=link}
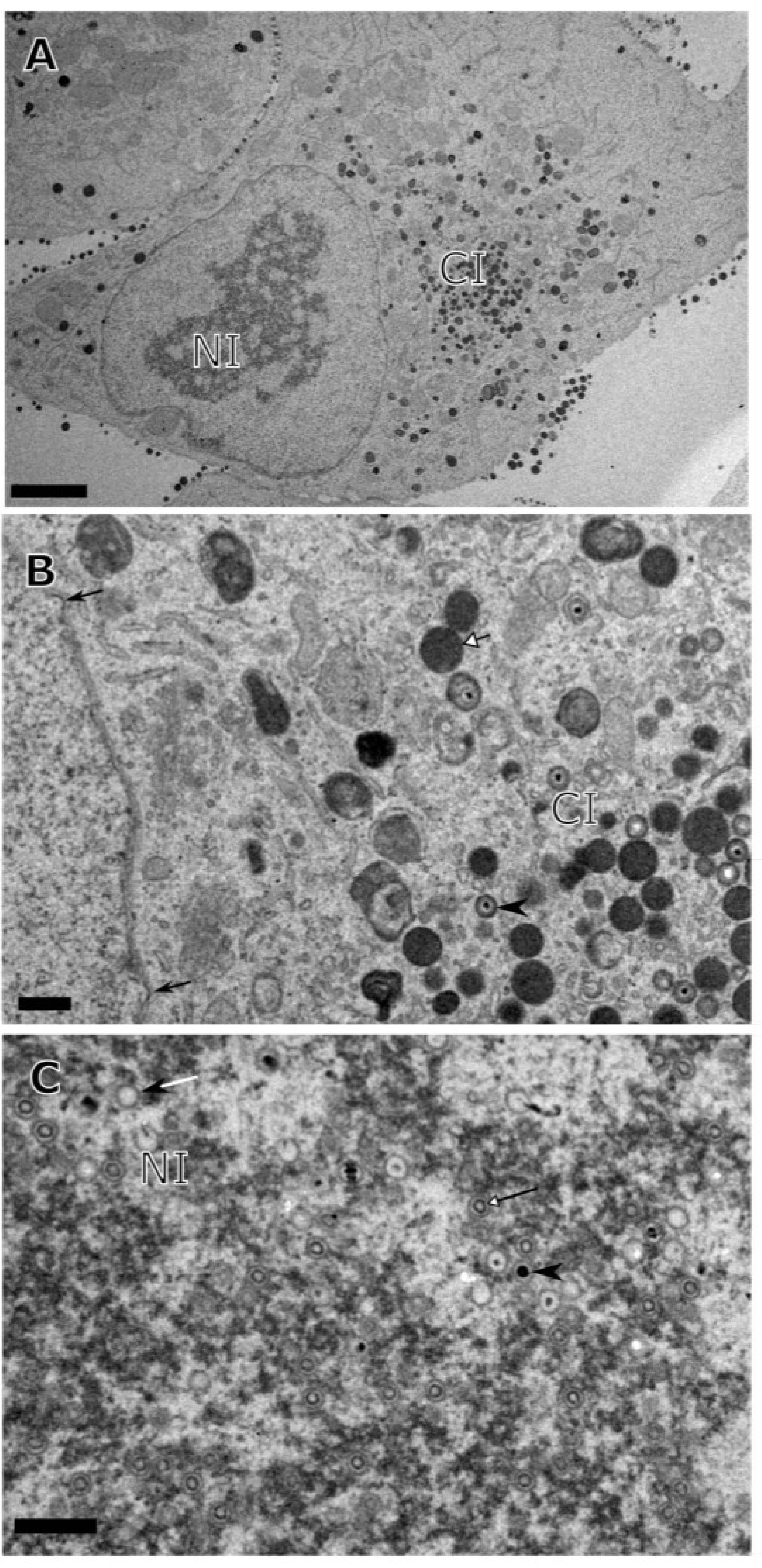{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of nuclear capsid | Mock-treated | BDCRB-treated | BDCRB release |
|---|---|---|---|
| A (% of total) | 27.30 | 10.75 | 25.00 |
| B (% of total) | 52.90 | 85.05 | 50.80 |
| C (% of total) | 19.83 | 4.20 | 23.40 |
| Average capsids per nucleus | 60.50 | 71.33 | 41.33 |
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, J.E. Virus particle maturation: Insights into elegantly programmed nanomachines. Curr. Opin. Struct. Biol. 2010, 20, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.H.; Grimes, J.M.; Stuart, D.I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.L. HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. USA 2014, 111, 6022–6027. [Google Scholar] [CrossRef] [PubMed]
- Selvarajan Sigamani, S.; Zhao, H.; Kamau, Y.N.; Baines, J.D.; Tang, L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 2013, 87, 7140–7148. [Google Scholar]
- Kondabagil, K.R.; Zhang, Z.; Rao, V.B. The DNA translocating ATPase of bacteriophage T4 packaging motor. J. Mol. Biol. 2006, 363, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Kottadiel, V.I.; Rao, V.B.; Chemla, Y.R. The dynamic pause-unpackaging state, an off-translocation recovery state of a DNA packaging motor from bacteriophage T4. Proc. Natl. Acad. Sci. USA 2012, 109, 20000–20005. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Juhas, R.M.; Thomsen, D.R.; Homa, F.L.; Burch, A.D.; Weller, S.K.; Brown, J.C. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 2001, 75, 10923–10932. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.W.; Huffman, J.B.; Homa, F.L.; Evilevitch, A. Herpes virus genome, the pressure is on. J. Am. Chem. Soc. 2013, 135, 11216–11221. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 2008, 325, 187–204. [Google Scholar] [PubMed]
- Britt, B. Maturation and egress. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Brown, J.C.; Newcomb, W.W. Herpesvirus capsid assembly: Insights from structural analysis. Curr. Opin. Virol. 2011, 1, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Cardone, G.; Winkler, D.C.; Trus, B.L.; Cheng, N.; Heuser, J.E.; Newcomb, W.W.; Brown, J.C.; Steven, A.C. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 2007, 361, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Huang, E.; Desai, J.; Sole, M.; Pryce, E.N.; Okoye, M.E.; Person, S.; Desai, P.J. A domain in the herpes simplex virus 1 triplex protein VP23 is essential for closure of capsid shells into icosahedral structures. J. Virol. 2011, 85, 12698–12707. [Google Scholar] [CrossRef] [PubMed]
- Trus, B.L.; Booy, F.P.; Newcomb, W.W.; Brown, J.C.; Homa, F.L.; Thomsen, D.R.; Steven, A.C. The herpes simplex virus procapsid: Structure, conformational changes upon maturation, and roles of the triplex proteins VP19c and VP23 in assembly. J. Mol. Biol. 1996, 263, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Homa, F.L.; Thomsen, D.R.; Trus, B.L.; Cheng, N.; Steven, A.; Booy, F.; Brown, J.C. Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes containing the major capsid and scaffolding proteins. J. Virol. 1999, 73, 4239–4250. [Google Scholar] [PubMed]
- Tandon, R.; Mocarski, E.S. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 2012, 20, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S., Jr.; Shenk, T.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 2701–2772. [Google Scholar]
- Albright, B.S.; Nellissery, J.; Szczepaniak, R.; Weller, S.K. Disulfide bond formation in the herpes simplex virus 1 UL6 protein is required for portal ring formation and genome encapsidation. J. Virol. 2011, 85, 8616–8624. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, R.; Nellissery, J.; Jadwin, J.A.; Makhov, A.M.; Kosinski, A.; Conway, J.F.; Weller, S.K. Disulfide bond formation contributes to herpes simplex virus capsid stability and retention of pentons. J. Virol. 2011, 85, 8625–8634. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Remillard-Labrosse, G.; Lippe, R. In vitro nuclear egress of herpes simplex virus type 1 capsids. Methods 2011, 55, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.; Roizman, B. Proteins specified by herpes simplex virus 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 1972, 10, 1044–1052. [Google Scholar] [PubMed]
- Yu, X.K.; O’Connor, C.M.; Atanasov, I.; Damania, B.; Kedes, D.H.; Zhou, Z.H. Three-dimensional structures of the A, B, and C capsids of rhesus monkey rhadinovirus: Insights into gammaherpesvirus capsid assembly, maturation, and DNA packaging. J. Virol. 2003, 77, 13182–13193. [Google Scholar] [CrossRef] [PubMed]
- Black, L.W.; Silverman, D.J. Model for DNA packaging into bacteriophage T4 heads. J. Virol. 1978, 28, 643–655. [Google Scholar] [PubMed]
- Laemmli, U.K.; Favre, M. Maturation of the head of bacteriophage T4 I. DNA packaging events. J. Mol. Biol. 1973, 80, 575–599. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D.N.; Raymer, D.M.; Rickgauer, J.P.; Robertson, R.M.; Catalano, C.E.; Anderson, D.L.; Grimes, S.; Smith, D.E. Measurements of single DNA molecule packaging dynamics in bacteriophage lambda reveal high forces, high motor processivity, and capsid transformations. J. Mol. Biol. 2007, 373, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D.N.; Raymer, D.M.; Kottadiel, V.I.; Rao, V.B.; Smith, D.E. Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc. Natl. Acad. Sci. USA 2007, 104, 16868–16873. [Google Scholar] [CrossRef] [PubMed]
- Rickgauer, J.P.; Fuller, D.N.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Smith, D.E. Portal motor velocity and internal force resisting viral DNA packaging in bacteriophage phi29. Biophys. J. 2008, 94, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J.; Boppana, S. Human cytomegalovirus virion proteins. Hum. Immunol. 2004, 65, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.R.; Harvey, R.J.; Stanat, S.C.; Hemphill, M.L.; Miller, T.; Drach, J.C.; Townsend, L.B.; Biron, K.K. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 1998, 72, 717–725. [Google Scholar] [PubMed]
- Thomsen, D.R.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Assembly of the herpes simplex virus capsid: Requirement for the carboxyl-terminal twenty-five amino acids of the proteins encoded by the UL26 and UL26.5 genes. J. Virol. 1995, 69, 3690–3703. [Google Scholar] [PubMed]
- Gibson, W. Protein counterparts of human and simian cytomegaloviruses. Virology 1983, 128, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Irmiere, A.; Gibson, W. Isolation of human cytomegalovirus intranuclear capsids, characterization of their protein constituents, and demonstration that the B-capsid assembly protein is also abundant in noninfectious enveloped particles. J. Virol. 1985, 56, 277–283. [Google Scholar] [PubMed]
- Yan, X.; Sinkovits, R.S.; Baker, T.S. AUTO3DEM—An automated and high throughput program for image reconstruction of icosahedral particles. J. Struct. Biol. 2007, 157, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pellett, P.E. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ortiz, D.A.; Gurczynski, S.J.; Khan, F.; Pellett, P.E. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J. Virol. 2014, 88, 9086–9099. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Mocarski, E.S. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J. Virol. 2008, 82, 9433–9444. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J.; Jarvis, M.; Seo, J.Y.; Drummond, D.; Nelson, J. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: Demonstration that pp28 (UL99) is essential for production of infectious virus. J. Virol. 2004, 78, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, V.; Greis, K.D.; Sztul, E.; Britt, W.J. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: Characterization of a potential site of virus assembly. J. Virol. 2000, 74, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, V.; Sztul, E.; Britt, W.J. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-golgi-intermediate compartment. J. Virol. 2000, 74, 3842–3851. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Yu, Q.C.; Enquist, L.; Shenk, T. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 2003, 77, 10594–10605. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Homa, F.L.; Thomsen, D.R.; Ye, Z.; Brown, J.C. Cell-free assembly of the herpes simplex virus capsid. J. Virol. 1994, 68, 6059–6063. [Google Scholar] [PubMed]
- Tatman, J.D.; Preston, V.G.; Nicholson, P.; Elliott, R.M.; Rixon, F.J. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J. Gen. Virol. 1994, 75, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Smith, G.B.; Meiering, C.D.; Mocarski, E.S. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J. Virol. 2006, 80, 8199–8210. [Google Scholar] [CrossRef] [PubMed]
- Trus, B.L.; Gibson, W.; Cheng, N.; Steven, A.C. Capsid structure of simian cytomegalovirus from cryoelectron microscopy: Evidence for tegument attachment sites. J. Virol. 1999, 73, 2181–2192. [Google Scholar] [PubMed]
- Evers, D.L.; Komazin, G.; Ptak, R.G.; Shin, D.; Emmer, B.T.; Townsend, L.B.; Drach, J.C. Inhibition of human cytomegalovirus replication by benzimidazole nucleosides involves three distinct mechanisms. Antimicrob. Agents Chemother. 2004, 48, 3918–3927. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, A.; Drach, J.C.; Townsend, L.B.; Fischer, A.; Bogner, E. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J. Virol. 2005, 79, 14660–14667. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shah, S.; Lee, M.; Dai, W.; Lo, P.; Britt, W.; Zhu, H.; Liu, F.; Zhou, Z.H. Biochemical and structural characterization of the capsid-bound tegument proteins of human cytomegalovirus. J. Struct. Biol. 2011, 174, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Krosky, P.M.; Underwood, M.R.; Turk, S.R.; Feng, K.W.; Jain, R.K.; Ptak, R.G.; Westerman, A.C.; Biron, K.K.; Townsend, L.B.; Drach, J.C.; et al. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 1998, 72, 4721–4728. [Google Scholar] [PubMed]
- Toropova, K.; Huffman, J.B.; Homa, F.L.; Conway, J.F. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J. Virol. 2011, 85, 7513–7522. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 2011, 85, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Mocarski, E.S. Cytomegalovirus pUL96 Is Critical for the Stability of pp150-Associated Nucleocapsids. J. Virol. 2011, 85, 7129–7141. [Google Scholar] [CrossRef] [PubMed]
- Cayatte, C.; Schneider-Ohrum, K.; Wang, Z.; Irrinki, A.; Nguyen, N.; Lu, J.; Nelson, C.; Servat, E.; Gemmell, L.; Citkowicz, A.; et al. Cytomegalovirus vaccine strain towne-derived dense bodies induce broad cellular immune responses and neutralizing antibodies that prevent infection of fibroblasts and epithelial cells. J. Virol. 2013, 87, 11107–11120. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tandon, R.; Mocarski, E.S.; Conway, J.F. The A, B, Cs of Herpesvirus Capsids. Viruses 2015, 7, 899-914. https://doi.org/10.3390/v7030899
Tandon R, Mocarski ES, Conway JF. The A, B, Cs of Herpesvirus Capsids. Viruses. 2015; 7(3):899-914. https://doi.org/10.3390/v7030899
Chicago/Turabian StyleTandon, Ritesh, Edward S. Mocarski, and James F. Conway. 2015. "The A, B, Cs of Herpesvirus Capsids" Viruses 7, no. 3: 899-914. https://doi.org/10.3390/v7030899
APA StyleTandon, R., Mocarski, E. S., & Conway, J. F. (2015). The A, B, Cs of Herpesvirus Capsids. Viruses, 7(3), 899-914. https://doi.org/10.3390/v7030899