B Cell Response and Mechanisms of Antibody Protection to West Nile Virus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virology and Pathogenesis
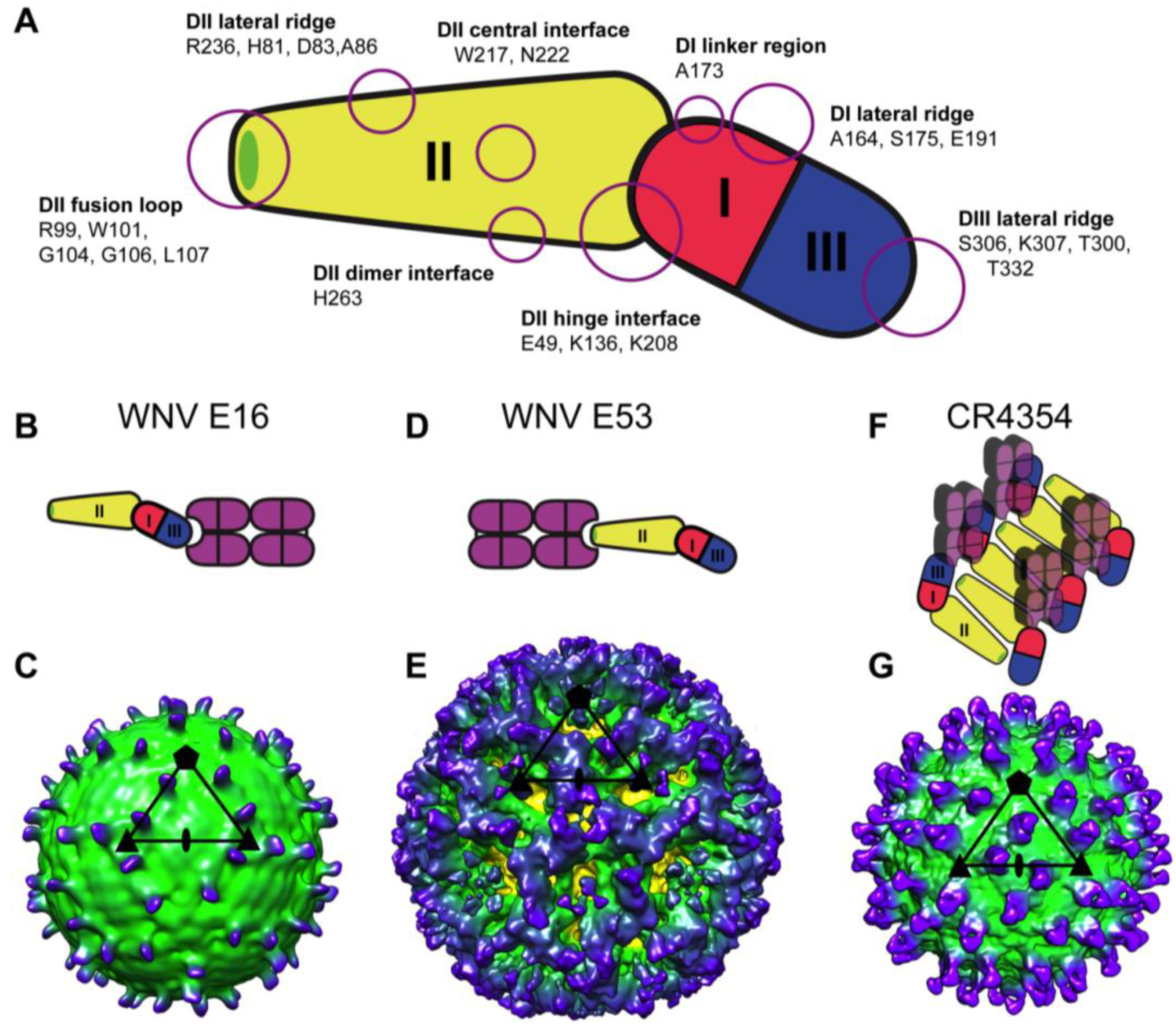
3. WNV Structural Biology

Structural Flexibility of WNV
4. Humoral Immune Response to WNV
4.1. Humoral Memory Response
4.2. Epitopes Targeted by WNV-Specific Antibodies

5. Mechanisms of WNV Neutralization
5.1. Antibody Affinity and Epitope Accessibility Govern WNV Neutralization
5.2. Factors That Modulate WNV Epitope Accessibility
5.2.1. Structural Heterogeneity of WNV due to Inefficient Maturation

5.2.2. Virus Breathing Increases Epitope Accessibility
6. Antibody Fc-Region Effector Functions
7. Understanding the Human Polyclonal Response to WNV
8. Progress of WNV Vaccine and Therapeutics
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Smithburn, K.C.; Hughes, T.P.; Burke, A.W.; Paul, J.H. A neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. Hyg. 1940, 20, 471–492. [Google Scholar]
- Centers for Disease Control and Prevention. West Nile virus and other arboviral diseases—United States, 2012. In MMWR Morb. Mortal. Wkly. Rep.; 2013; 62, pp. 513–517. [Google Scholar]
- Mukhopadhyay, S.; Kim, B.S.; Chipman, P.R.; Rossmann, M.G.; Kuhn, R.J. Structure of West Nile virus. Science 2003, 302, e248. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 2004, 279, 54533–54541. [Google Scholar] [CrossRef]
- Lee, J.W.; Chu, J.J.; Ng, M.L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar] [CrossRef]
- Davis, C.W.; Mattei, L.M.; Nguyen, H.Y.; Ansarah-Sobrinho, C.; Doms, R.W.; Pierson, T.C. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J. Biol. Chem. 2006, 281, 37183–37194. [Google Scholar]
- Davis, C.W.; Nguyen, H.Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef]
- Hershkovitz, O.; Rosental, B.; Rosenberg, L.A.; Navarro-Sanchez, M.E.; Jivov, S.; Zilka, A.; Gershoni-Yahalom, O.; Brient-Litzler, E.; Bedouelle, H.; Ho, J.W.; et al. NKp44 receptor mediates interaction of the envelope glycoproteins from the West Nile and dengue viruses with NK cells. J. Immunol. 2009, 183, 2610–2621. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 2004, 78, 10543–10555. [Google Scholar] [CrossRef]
- Gollins, S.W.; Porterfield, J.S. Flavivirus infection enhancement in macrophages: Radioactive and biological studies on the effect of antibody on viral fate. J. Gen. Virol. 1984, 65, 1261–1272. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K.; Allison, S.L. The entry machinery of flaviviruses. Arch. Virol. Suppl. 2004, 18, 133–137. [Google Scholar]
- Van der Schaar, H.M.; Rust, M.J.; Waarts, B.L.; van der Ende-Metselaar, H.; Kuhn, R.J.; Wilschut, J.; Zhuang, X.; Smit, J.M. Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol. 2007, 81, 12019–12028. [Google Scholar] [CrossRef]
- Cockburn, J.J.; Navarro Sanchez, M.E.; Goncalvez, A.P.; Zaitseva, E.; Stura, E.A.; Kikuti, C.M.; Duquerroy, S.; Dussart, P.; Chernomordik, L.V.; Lai, C.J.; et al. Structural insights into the neutralization mechanism of a higher primate antibody against dengue virus. EMBO J. 2012, 31, 767–779. [Google Scholar] [CrossRef]
- Kanai, R.; Kar, K.; Anthony, K.; Gould, L.H.; Ledizet, M.; Fikrig, E.; Marasco, W.A.; Koski, R.A.; Modis, Y. Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J. Virol. 2006, 80, 11000–11008. [Google Scholar] [CrossRef]
- Luca, V.C.; AbiMansour, J.; Nelson, C.A.; Fremont, D.H. Crystal structure of the Japanese encephalitis virus envelope protein. J. Virol. 2012, 86, 2337–2346. [Google Scholar] [CrossRef]
- Luca, V.C.; Nelson, C.A.; Fremont, D.H. Structure of the St. Louis encephalitis virus postfusion envelope trimer. J. Virol. 2013, 87, 818–828. [Google Scholar] [CrossRef]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef]
- Nayak, V.; Dessau, M.; Kucera, K.; Anthony, K.; Ledizet, M.; Modis, Y. Crystal structure of dengue virus type 1 envelope protein in the postfusion conformation and its implications for membrane fusion. J. Virol. 2009, 83, 4338–4344. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Nelson, C.A.; Chen, B.R.; Diamond, M.S.; Fremont, D.H. Crystal structure of the West Nile virus envelope glycoprotein. J. Virol. 2006, 80, 11467–11474. [Google Scholar] [CrossRef]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Zhang, W.; Kaufmann, B.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G. Membrane curvature in flaviviruses. J. Struct. Biol. 2013, 183, 86–94. [Google Scholar] [CrossRef]
- Zhang, X.; Ge, P.; Yu, X.; Brannan, J.M.; Bi, G.; Zhang, Q.; Schein, S.; Zhou, Z.H. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat. Struct. Mol. Biol. 2013, 20, 105–110. [Google Scholar]
- Oliphant, T.; Nybakken, G.E.; Engle, M.; Xu, Q.; Nelson, C.A.; Sukupolvi-Petty, S.; Marri, A.; Lachmi, B.E.; Olshevsky, U.; Fremont, D.H.; et al. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J. Virol. 2006, 80, 12149–12159. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.3r1; Schrodinger, LLC: New York, NY, USA, 2010.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Zhang, Y.; Kaufmann, B.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G. Structure of immature West Nile virus. J. Virol. 2007, 81, 6141–6145. [Google Scholar] [CrossRef]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef]
- Moesker, B.; Rodenhuis-Zybert, I.A.; Meijerhof, T.; Wilschut, J.; Smit, J.M. Characterization of the functional requirements of West Nile virus membrane fusion. J. Gen. Virol. 2010, 91, 389–393. [Google Scholar] [CrossRef]
- Nelson, S.; Jost, C.A.; Xu, Q.; Ess, J.; Martin, J.E.; Oliphant, T.; Whitehead, S.S.; Durbin, A.P.; Graham, B.S.; Diamond, M.S.; et al. Maturation of West Nile virus modulates sensitivity to antibody-mediated neutralization. PLoS Pathog. 2008, 4, e1000060. [Google Scholar] [CrossRef]
- Vogt, M.R.; Dowd, K.A.; Engle, M.; Tesh, R.B.; Johnson, S.; Pierson, T.C.; Diamond, M.S. Poorly neutralizing cross-reactive antibodies against the fusion loop of West Nile virus envelope protein protect in vivo via Fcgamma receptor and complement-dependent effector mechanisms. J. Virol. 2011, 85, 11567–11580. [Google Scholar] [CrossRef]
- Stiasny, K.; Kiermayr, S.; Bernhart, A.; Heinz, F.X. The membrane-proximal “stem” region increases the stability of the flavivirus e protein postfusion trimer and modulates its structure. J. Virol. 2013, 87, 9933–9938. [Google Scholar] [CrossRef]
- Fibriansah, G.; Ng, T.S.; Kostyuchenko, V.A.; Lee, J.; Lee, S.; Wang, J.; Lok, S.M. Structural changes in dengue virus when exposed to a temperature of 37 degrees C. J. Virol. 2013, 87, 7585–7592. [Google Scholar] [CrossRef]
- Zhang, X.; Sheng, J.; Plevka, P.; Kuhn, R.J.; Diamond, M.S.; Rossmann, M.G. Dengue structure differs at the temperatures of its human and mosquito hosts. Proc. Natl. Acad. Sci. USA 2013, 110, 6795–6799. [Google Scholar] [CrossRef]
- Austin, S.K.; Dowd, K.A.; Shrestha, B.; Nelson, C.A.; Edeling, M.A.; Johnson, S.; Pierson, T.C.; Diamond, M.S.; Fremont, D.H. Structural basis of differential neutralization of DENV-1 genotypes by an antibody that recognizes a cryptic epitope. PLoS Pathog. 2012, 8, e1002930. [Google Scholar] [CrossRef]
- Sukupolvi-Petty, S.; Brien, J.D.; Austin, S.K.; Shrestha, B.; Swayne, S.; Kahle, K.; Doranz, B.J.; Johnson, S.; Pierson, T.C.; Fremont, D.H.; et al. Functional analysis of antibodies against dengue virus type 4 reveals strain-dependent epitope exposure that impacts neutralization and protection. J. Virol. 2013, 87, 8826–8842. [Google Scholar] [CrossRef]
- Dowd, K.A.; Jost, C.A.; Durbin, A.P.; Whitehead, S.S.; Pierson, T.C. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog. 2011, 7, e1002111. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Engle, M.J.; Diamond, M.S. Antibody prophylaxis and therapy against West Nile virus infection in wild-type and immunodeficient mice. J. Virol. 2003, 77, 12941–12949. [Google Scholar] [CrossRef]
- Purtha, W.E.; Chachu, K.A.; Virgin, H.W.T.; Diamond, M.S. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J. Virol. 2008, 82, 10964–10974. [Google Scholar] [CrossRef]
- Xia, J.; Winkelmann, E.R.; Gorder, S.R.; Mason, P.W.; Milligan, G.N. TLR3- and MyD88-dependent signaling differentially influence the development of West Nile virus-specific B cell responses in mice following immunization with the single-cycle flavivirus (SCFV) RepliVAX WN. J. Virol. 2013, 87, 12090–12101. [Google Scholar] [CrossRef]
- Stewart, B.S.; Demarest, V.L.; Wong, S.J.; Green, S.; Bernard, K.A. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 2011, 12, e6. [Google Scholar] [CrossRef]
- Ma, D.Y.; Suthar, M.S.; Kasahara, S.; Gale, M., Jr.; Clark, E.A. CD22 is required for protection against West Nile virus Infection. J. Virol. 2013, 87, 3361–3375. [Google Scholar] [CrossRef]
- Mehlhop, E.; Ansarah-Sobrinho, C.; Johnson, S.; Engle, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an IgG subclass-specific manner. Cell Host Microbe 2007, 2, 417–426. [Google Scholar] [CrossRef]
- Mehlhop, E.; Diamond, M.S. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006, 203, 1371–1381. [Google Scholar] [CrossRef]
- Sitati, E.; McCandless, E.E.; Klein, R.S.; Diamond, M.S. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J. Virol. 2007, 81, 9801–9811. [Google Scholar] [CrossRef]
- Sitati, E.M.; Diamond, M.S. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef]
- Sabin, A.B. Research on dengue during World War II. Am. J. Trop. Med. Hyg. 1952, 1, 30–50. [Google Scholar]
- Chabierski, S.; Makert, G.R.; Kerzhner, A.; Barzon, L.; Fiebig, P.; Liebert, U.G.; Papa, A.; Richner, J.M.; Niedrig, M.; Diamond, M.S.; et al. Antibody responses in humans infected with newly emerging strains of West Nile Virus in Europe. PLoS One 2013, 8, e66507. [Google Scholar]
- Oliphant, T.; Engle, M.; Nybakken, G.E.; Doane, C.; Johnson, S.; Huang, L.; Gorlatov, S.; Mehlhop, E.; Marri, A.; Chung, K.M.; et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005, 11, 522–530. [Google Scholar] [CrossRef]
- Throsby, M.; Geuijen, C.; Goudsmit, J.; Bakker, A.Q.; Korimbocus, J.; Kramer, R.A.; Clijsters-van der Horst, M.; de Jong, M.; Jongeneelen, M.; Thijsse, S.; et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J. Virol. 2006, 80, 6982–6992. [Google Scholar] [CrossRef]
- Ledizet, M.; Kar, K.; Foellmer, H.G.; Wang, T.; Bushmich, S.L.; Anderson, J.F.; Fikrig, E.; Koski, R.A. A recombinant envelope protein vaccine against West Nile virus. Vaccine 2005, 23, 3915–3924. [Google Scholar] [CrossRef]
- Pinto, A.K.; Richner, J.M.; Poore, E.A.; Patil, P.P.; Amanna, I.J.; Slifka, M.K.; Diamond, M.S. A hydrogen peroxide-inactivated virus vaccine elicits humoral and cellular immunity and protects against lethal West Nile virus infection in aged mice. J. Virol. 2013, 87, 1926–1936. [Google Scholar] [CrossRef]
- Tesh, R.B.; Arroyo, J.; Travassos Da Rosa, A.P.; Guzman, H.; Xiao, S.Y.; Monath, T.P. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerging Infect. Dis. 2002, 8, 1392–1397. [Google Scholar] [CrossRef]
- Purtha, W.E.; Tedder, T.F.; Johnson, S.; Bhattacharya, D.; Diamond, M.S. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J. Exp. Med. 2011, 208, 2599–2606. [Google Scholar] [CrossRef]
- Petrovsky, N.; Larena, M.; Siddharthan, V.; Prow, N.A.; Hall, R.A.; Lobigs, M.; Morrey, J. An inactivated cell culture Japanese encephalitis vaccine (JE-ADVAX) formulated with delta inulin adjuvant provides robust heterologous protection against West Nile encephalitis via cross-protective memory B cells and neutralizing antibody. J. Virol. 2013, 87, 10324–10333. [Google Scholar] [CrossRef]
- Sultana, H.; Foellmer, H.G.; Neelakanta, G.; Oliphant, T.; Engle, M.; Ledizet, M.; Krishnan, M.N.; Bonafe, N.; Anthony, K.G.; Marasco, W.A.; et al. Fusion loop peptide of the West Nile virus envelope protein is essential for pathogenesis and is recognized by a therapeutic cross-reactive human monoclonal antibody. J. Immunol. 2009, 183, 650–660. [Google Scholar] [CrossRef]
- Gould, L.H.; Sui, J.; Foellmer, H.; Oliphant, T.; Wang, T.; Ledizet, M.; Murakami, A.; Noonan, K.; Lambeth, C.; Kar, K.; et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J. Virol. 2005, 79, 14606–14613. [Google Scholar] [CrossRef]
- Cherrier, M.V.; Kaufmann, B.; Nybakken, G.E.; Lok, S.M.; Warren, J.T.; Chen, B.R.; Nelson, C.A.; Kostyuchenko, V.A.; Holdaway, H.A.; Chipman, P.R.; et al. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 2009, 28, 3269–3276. [Google Scholar] [CrossRef]
- Vogt, M.R.; Moesker, B.; Goudsmit, J.; Jongeneelen, M.; Austin, S.K.; Oliphant, T.; Nelson, S.; Pierson, T.C.; Wilschut, J.; Throsby, M.; et al. Human monoclonal antibodies against West Nile virus induced by natural infection neutralize at a postattachment step. J. Virol. 2009, 83, 6494–6507. [Google Scholar] [CrossRef]
- Oliphant, T.; Nybakken, G.E.; Austin, S.K.; Xu, Q.; Bramson, J.; Loeb, M.; Throsby, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Induction of epitope-specific neutralizing antibodies against West Nile virus. J. Virol. 2007, 81, 11828–11839. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Oliphant, T.; Johnson, S.; Burke, S.; Diamond, M.S.; Fremont, D.H. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 2005, 437, 764–769. [Google Scholar] [CrossRef]
- Sanchez, M.D.; Pierson, T.C.; McAllister, D.; Hanna, S.L.; Puffer, B.A.; Valentine, L.E.; Murtadha, M.M.; Hoxie, J.A.; Doms, R.W. Characterization of neutralizing antibodies to West Nile virus. Virology 2005, 336, 70–82. [Google Scholar] [CrossRef]
- Pierson, T.C.; Xu, Q.; Nelson, S.; Oliphant, T.; Nybakken, G.E.; Fremont, D.H.; Diamond, M.S. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 2007, 1, 135–145. [Google Scholar] [CrossRef]
- Diamond, M.S.; Pierson, T.C.; Fremont, D.H. The structural immunology of antibody protection against West Nile virus. Immunol. Rev. 2008, 225, 212–225. [Google Scholar] [CrossRef]
- Kaufmann, B.; Vogt, M.R.; Goudsmit, J.; Holdaway, H.A.; Aksyuk, A.A.; Chipman, P.R.; Kuhn, R.J.; Diamond, M.S.; Rossmann, M.G. Neutralization of West Nile virus by cross-linking of its surface proteins with Fab fragments of the human monoclonal antibody CR4354. Proc. Natl. Acad. Sci. USA 2010, 107, 18950–18955. [Google Scholar] [CrossRef]
- Kaufmann, B.; Nybakken, G.E.; Chipman, P.R.; Zhang, W.; Diamond, M.S.; Fremont, D.H.; Kuhn, R.J.; Rossmann, M.G. West Nile virus in complex with the Fab fragment of a neutralizing monoclonal antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 12400–12404. [Google Scholar] [CrossRef]
- Faggioni, G.; Pomponi, A.; de Santis, R.; Masuelli, L.; Ciammaruconi, A.; Monaco, F.; di Gennaro, A.; Marzocchella, L.; Sambri, V.; Lelli, R.; et al. West Nile alternative open reading frame (N-NS4B/WARF4) is produced in infected West Nile Virus (WNV) cells and induces humoral response in WNV infected individuals. Virol. J. 2012, 9, e283. [Google Scholar] [CrossRef]
- Calvert, A.E.; Kalantarov, G.F.; Chang, G.J.; Trakht, I.; Blair, C.D.; Roehrig, J.T. Human monoclonal antibodies to West Nile virus identify epitopes on the prM protein. Virology 2011, 410, 30–37. [Google Scholar] [CrossRef]
- Hirota, J.; Shimizu, S. A new competitive ELISA detects West Nile virus infection using monoclonal antibodies against the precursor-membrane protein of West Nile virus. J. Virol. Methods 2013, 188, 132–138. [Google Scholar] [CrossRef]
- Chung, K.M.; Nybakken, G.E.; Thompson, B.S.; Engle, M.J.; Marri, A.; Fremont, D.H.; Diamond, M.S. Antibodies against West Nile virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 2006, 80, 1340–1351. [Google Scholar] [CrossRef]
- Chung, K.M.; Thompson, B.S.; Fremont, D.H.; Diamond, M.S. Antibody recognition of cell surface-associated NS1 triggers Fc-gamma receptor-mediated phagocytosis and clearance of West Nile Virus-infected cells. J. Virol. 2007, 81, 9551–9555. [Google Scholar] [CrossRef]
- Wong, S.J.; Boyle, R.H.; Demarest, V.L.; Woodmansee, A.N.; Kramer, L.D.; Li, H.; Drebot, M.; Koski, R.A.; Fikrig, E.; Martin, D.A.; et al. Immunoassay targeting nonstructural protein 5 to differentiate West Nile virus infection from dengue and St. Louis encephalitis virus infections and from flavivirus vaccination. J. Clin. Microbiol. 2003, 41, 4217–4223. [Google Scholar] [CrossRef]
- Dowd, K.A.; Pierson, T.C. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology 2011, 411, 306–315. [Google Scholar] [CrossRef]
- Mehlhop, E.; Nelson, S.; Jost, C.A.; Gorlatov, S.; Johnson, S.; Fremont, D.H.; Diamond, M.S.; Pierson, T.C. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe 2009, 6, 381–391. [Google Scholar] [CrossRef]
- Burton, D.R.; Saphire, E.O.; Parren, P.W. A model for neutralization of viruses based on antibody coating of the virion surface. Curr. Top. Microbiol. Immunol. 2001, 260, 109–143. [Google Scholar]
- Della-Porta, A.J.; Westaway, E.G. A multi-hit model for the neutralization of animal viruses. J. Gen. Virol. 1978, 38, 1–19. [Google Scholar] [CrossRef]
- Kaufmann, B.; Chipman, P.R.; Holdaway, H.A.; Johnson, S.; Fremont, D.H.; Kuhn, R.J.; Diamond, M.S.; Rossmann, M.G. Capturing a flavivirus pre-fusion intermediate. PLoS Pathog. 2009, 5, e1000672. [Google Scholar] [CrossRef]
- Thompson, B.S.; Moesker, B.; Smit, J.M.; Wilschut, J.; Diamond, M.S.; Fremont, D.H. A therapeutic antibody against West Nile virus neutralizes infection by blocking fusion within endosomes. PLoS Pathog. 2009, 5, e1000453. [Google Scholar] [CrossRef]
- Edeling, M.A.; Austin, S.K.; Dowd, K.A.; Pierson, T.C.; Diamond, M.S.; Fremont, D.H.; Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA. Unpublished work. 2014.
- Klasse, P.J.; Sattentau, Q.J. Mechanisms of virus neutralization by antibody. Curr. Top. Microbiol. Immunol. 2001, 260, 87–108. [Google Scholar]
- Zhang, S.; Vogt, M.R.; Oliphant, T.; Engle, M.; Bovshik, E.I.; Diamond, M.S.; Beasley, D.W. Development of resistance to passive therapy with a potently neutralizing humanized monoclonal antibody against West Nile virus. J. Infect. Dis. 2009, 200, 202–205. [Google Scholar] [CrossRef]
- Mukherjee, S.; Lin, T.Y.; Dowd, K.A.; Manhart, C.J.; Pierson, T.C. The infectivity of prM-containing partially mature West Nile virus does not require the activity of cellular furin-like proteases. J. Virol. 2011, 85, 12067–12072. [Google Scholar] [CrossRef]
- Lee, P.D.; Mukherjee, S.; Edeling, M.A.; Dowd, K.A.; Austin, S.K.; Manhart, C.J.; Diamond, M.S.; Fremont, D.; Pierson, T.C. The Fc region of an antibody impacts the neutralization of West Nile viruses in different maturation states. J. Virol. 2013, 24, 13729–13740. [Google Scholar]
- Dowd, K.A.; Mukherjee, S.; Pierson, T.C.; Laboratory of Viral Diseases, National Institute for Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA. Unpublished work. 2014.
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2008, 6, 132–142. [Google Scholar] [CrossRef]
- Fuchs, A.; Lin, T.Y.; Beasley, D.W.; Stover, C.M.; Schwaeble, W.J.; Pierson, T.C.; Diamond, M.S. Direct complement restriction of flavivirus infection requires glycan recognition by mannose-binding lectin. Cell Host Microbe 2010, 8, 186–195. [Google Scholar] [CrossRef]
- Fuchs, A.; Pinto, A.K.; Schwaeble, W.J.; Diamond, M.S. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 2011, 412, 101–109. [Google Scholar] [CrossRef]
- Porterfield, J.S. Antibody-dependent enhancement of viral infectivity. Adv. Virus Res. 1986, 31, 335–355. [Google Scholar] [CrossRef]
- Murphy, B.R.; Whitehead, S.S. Immune response to dengue virus and prospects for a vaccine. Annu. Rev. Immunol. 2011, 29, 587–619. [Google Scholar] [CrossRef]
- Peiris, J.S.; Porterfield, J.S. Antibody-mediated enhancement of Flavivirus replication in macrophage-like cell lines. Nature 1979, 282, 509–511. [Google Scholar] [CrossRef]
- Pierson, T.C.; Sanchez, M.D.; Puffer, B.A.; Ahmed, A.A.; Geiss, B.J.; Valentine, L.E.; Altamura, L.A.; Diamond, M.S.; Doms, R.W. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 2006, 346, 53–65. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Rodenhuis-Zybert, I.; Moesker, B.; Wang, P.; Fikrig, E.; Smit, J.M. prM-antibody renders immature West Nile virus infectious in vivo. J. Gen. Virol. 2011, 92, 2281–2285. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.A.; Moesker, B.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Diamond, M.S.; Wilschut, J.; Smit, J.M. A fusion-loop antibody enhances the infectious properties of immature flavivirus particles. J. Virol. 2011, 85, 11800–11808. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.A.; van der Schaar, H.M.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Lei, H.Y.; Wilschut, J.; Smit, J.M. Immature dengue virus: A veiled pathogen? PLoS Pathog. 2010, 6, e1000718. [Google Scholar] [CrossRef]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–130100. [Google Scholar] [CrossRef]
- Chambers, T.J.; Halevy, M.; Nestorowicz, A.; Rice, C.M.; Lustig, S. West Nile virus envelope proteins: Nucleotide sequence analysis of strains differing in mouse neuroinvasiveness. J. Gen. Virol. 1998, 79, 2375–2380. [Google Scholar]
- Volk, D.E.; Beasley, D.W.; Kallick, D.A.; Holbrook, M.R.; Barrett, A.D.; Gorenstein, D.G. Solution structure and antibody binding studies of the envelope protein domain III from the New York strain of West Nile virus. J. Biol. Chem. 2004, 279, 38755–38761. [Google Scholar]
- Wahala, W.M.; Kraus, A.A.; Haymore, L.B.; Accavitti-Loper, M.A.; de Silva, A.M. Dengue virus neutralization by human immune sera: Role of envelope protein domain III-reactive antibody. Virology 2009, 392, 103–113. [Google Scholar] [CrossRef]
- De Alwis, R.; Smith, S.A.; Olivarez, N.P.; Messer, W.B.; Huynh, J.P.; Wahala, W.M.; White, L.J.; Diamond, M.S.; Baric, R.S.; Crowe, J.E., Jr.; et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl. Acad. Sci. USA 2012, 109, 7439–7444. [Google Scholar] [CrossRef]
- Beasley, D.W. Vaccines and immunotherapeutics for the prevention and treatment of infections with West Nile virus. Immunotherapy 2011, 3, 269–285. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Gershoni-Yahalom, O.; Samina, I.; Khinich, Y.; Nur, I.; Laub, O.; Gottreich, A.; Simanov, M.; Porgador, A.; Rager-Zisman, B.; et al. Using high titer West Nile intravenous immunoglobulin from selected Israeli donors for treatment of West Nile virus infection. BMC Infect. Dis. 2009, 9, e18. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Lustig, S.; Tam, G.; Robinzon, S.; Segal, S.; Rager-Zisman, B. Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating West Nile virus infection in mice. J. Infect. Dis. 2003, 188, 5–12. [Google Scholar] [CrossRef]
- Planitzer, C.B.; Modrof, J.; Kreil, T.R. West Nile virus neutralization by US plasma-derived immunoglobulin products. J. Infect. Dis. 2007, 196, 435–440. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. West Nile virus infections in organ transplant recipients—New York and Pennsylvania, August-September, 2005. In MMWR Morb. Mortal. Wkly. Rep.; 2005; 54, pp. 1021–1023. [Google Scholar]
- Centers for Disease Control and Prevention. West Nile virus transmission via organ transplantation and blood transfusion—Louisiana, 2008. In MMWR Morb. Mortal. Wkly. Rep.; 2009; 58, pp. 1263–1267. [Google Scholar]
- Iwamoto, M.; Jernigan, D.B.; Guasch, A.; Trepka, M.J.; Blackmore, C.G.; Hellinger, W.C.; Pham, S.M.; Zaki, S.; Lanciotti, R.S.; Lance-Parker, S.E.; et al. West Nile virus in transplant recipients investigation. Transmission of West Nile virus from an organ donor to four transplant recipients. N. Engl. J. Med. 2003, 348, 2196–2203. [Google Scholar] [CrossRef]
- Morelli, M.C.; Sambri, V.; Grazi, G.L.; Gaibani, P.; Pierro, A.; Cescon, M.; Ercolani, G.; Cavrini, F.; Rossini, G.; Capobianchi, M.R.; et al. Absence of neuroinvasive disease in a liver transplant recipient who acquired West Nile virus (WNV) infection from the organ donor and who received WNV antibodies prophylactically. Clin. Infect. Dis. 2010, 51, e34–e37. [Google Scholar] [CrossRef]
- Rhee, C.; Eaton, E.F.; Concepcion, W.; Blackburn, B.G. West Nile virus encephalitis acquired via liver transplantation and clinical response to intravenous immunoglobulin: Case report and review of the literature. Transpl. Infect. Dis. 2011, 13, 312–317. [Google Scholar] [CrossRef]
- Saquib, R.; Randall, H.; Chandrakantan, A.; Spak, C.W.; Barri, Y.M. West Nile virus encephalitis in a renal transplant recipient: The role of intravenous immunoglobulin. Am. J. Kidney Dis. 2008, 52, e19–e21. [Google Scholar] [CrossRef]
- National Institutes of Health Clinical Center (CC). Omr-IgG-am(Trademark) for Treating Patients with or at High Risk for West Nile Virus Disease. Available online: http://clinicaltrials.gov/ct2/show/record/NCT00069316/ (accessed on 15 September 2013).
- National Institutes of Health Clinical Center (CC). Treatment of West Nile Virus with MGAWN1 (PARADIGM). Available online: http://clinicaltrials.gov/ct2/show/record/NCT00927953/ (accessed on 15 September 2013).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Austin, S.K.; Dowd, K.A. B Cell Response and Mechanisms of Antibody Protection to West Nile Virus. Viruses 2014, 6, 1015-1036. https://doi.org/10.3390/v6031015
Austin SK, Dowd KA. B Cell Response and Mechanisms of Antibody Protection to West Nile Virus. Viruses. 2014; 6(3):1015-1036. https://doi.org/10.3390/v6031015
Chicago/Turabian StyleAustin, S. Kyle, and Kimberly A. Dowd. 2014. "B Cell Response and Mechanisms of Antibody Protection to West Nile Virus" Viruses 6, no. 3: 1015-1036. https://doi.org/10.3390/v6031015
APA StyleAustin, S. K., & Dowd, K. A. (2014). B Cell Response and Mechanisms of Antibody Protection to West Nile Virus. Viruses, 6(3), 1015-1036. https://doi.org/10.3390/v6031015