
Molecular Biology of KSHV Lytic Reactivation



Abstract
:1. Introduction
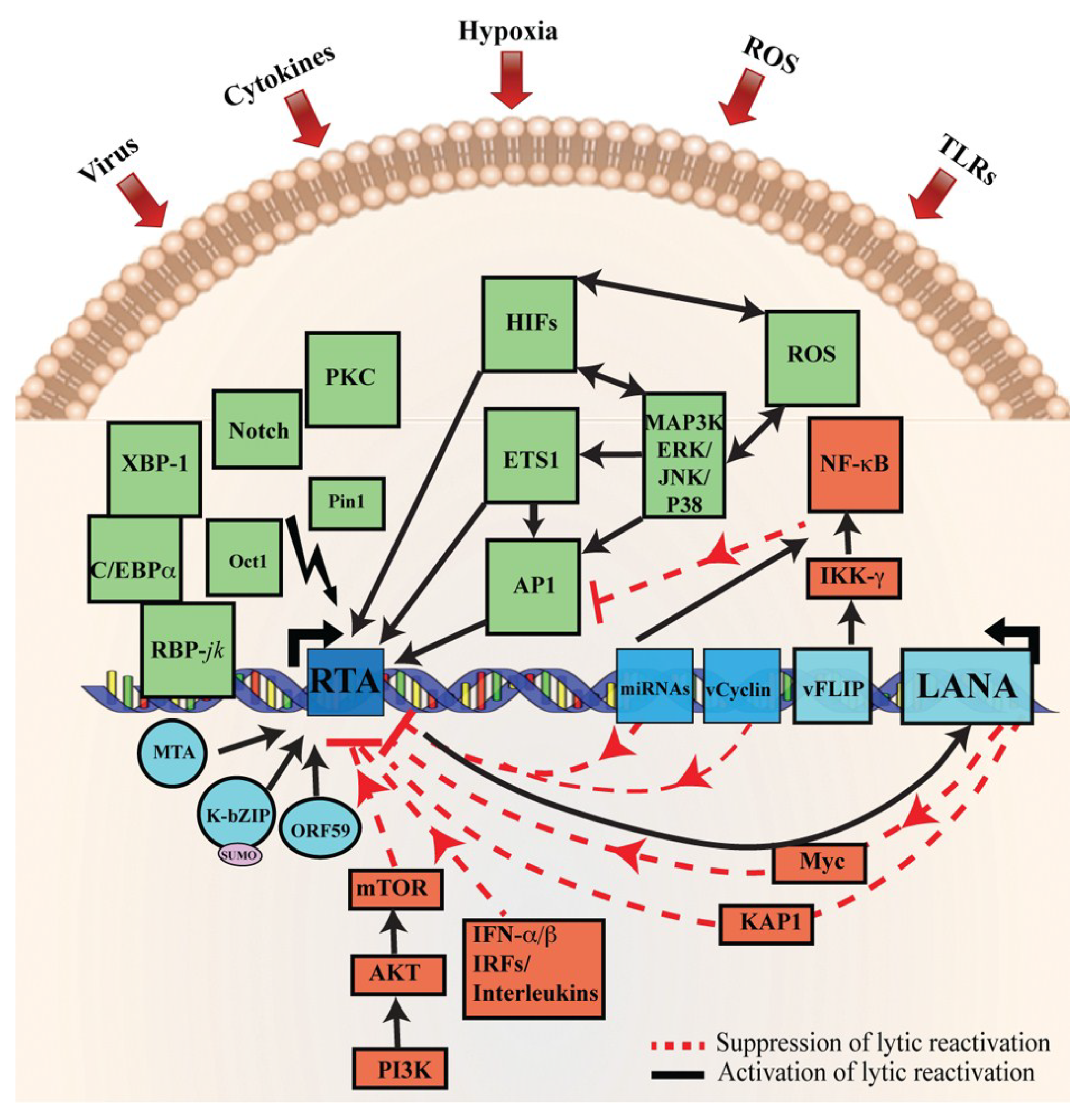
2. LANA and KSHV Reactivation
3. Stimulus Triggering KSHV Reactivation
3.1. Viral Co-Infection
3.2. Hypoxia
3.3. Oxidative Stress and Reactive Oxygen Species (ROS)
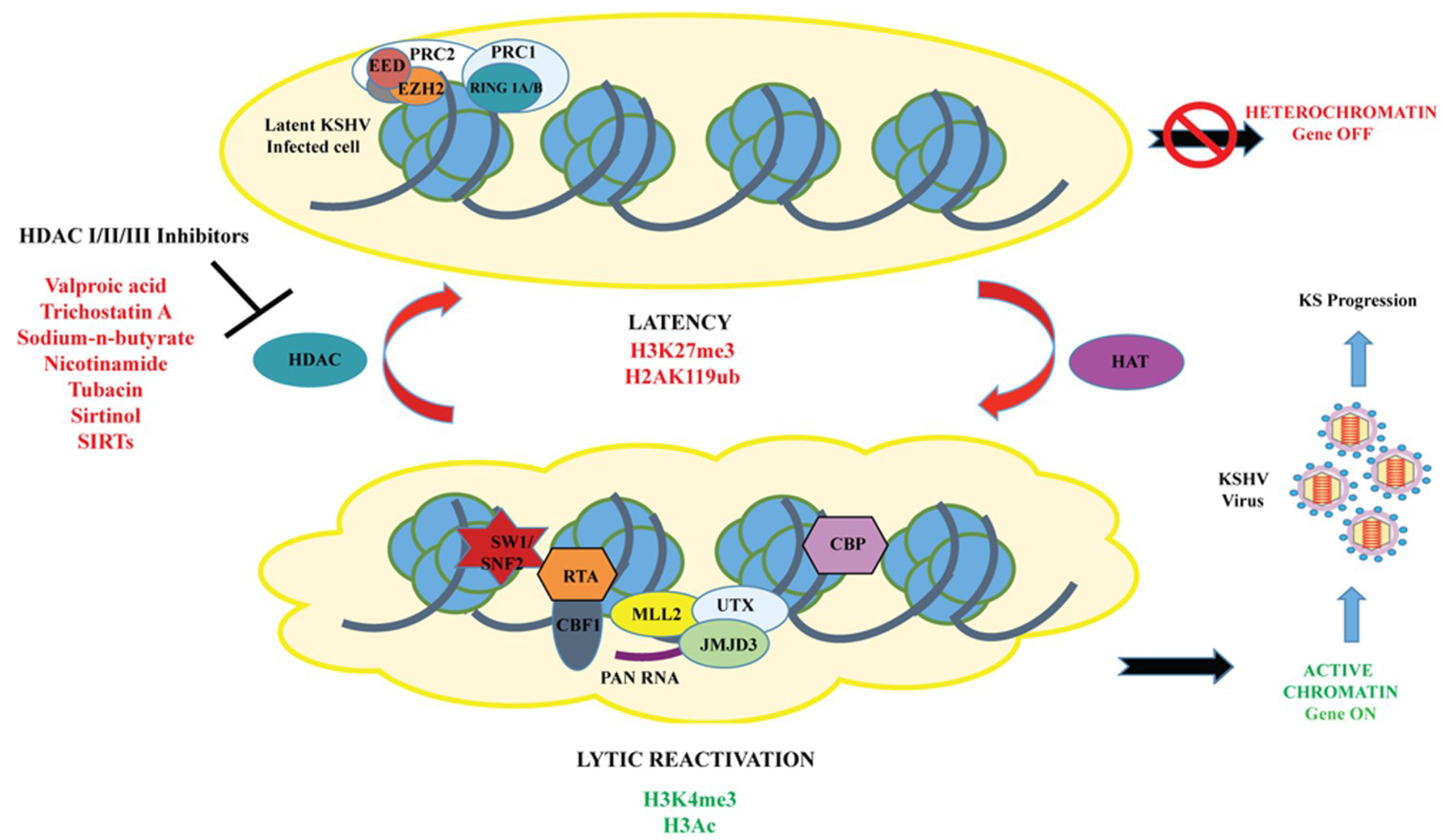
3.4. Histone Deacetylases and Histone Deacetylase Inhibitors (HDACs and HDACi)

3.5. Dietary Supplements
4. Role of Viral and Cellular Proteins Important for Lytic DNA Replication
4.1. Viral Factors
4.1.1. K-RTA (KSHV Replication and Transcription Activator)
4.1.2. ORF57-mRNA Transcript Accumulation (MTA)
4.1.3. KSHV K8-K-bZIP—Lytic Replication-Associated Protein (RAP)
4.1.4. ORF59- Viral Processivity Factor
4.1.5. ORF6-Single Strand Binding Protein
4.2. Cellular Factors

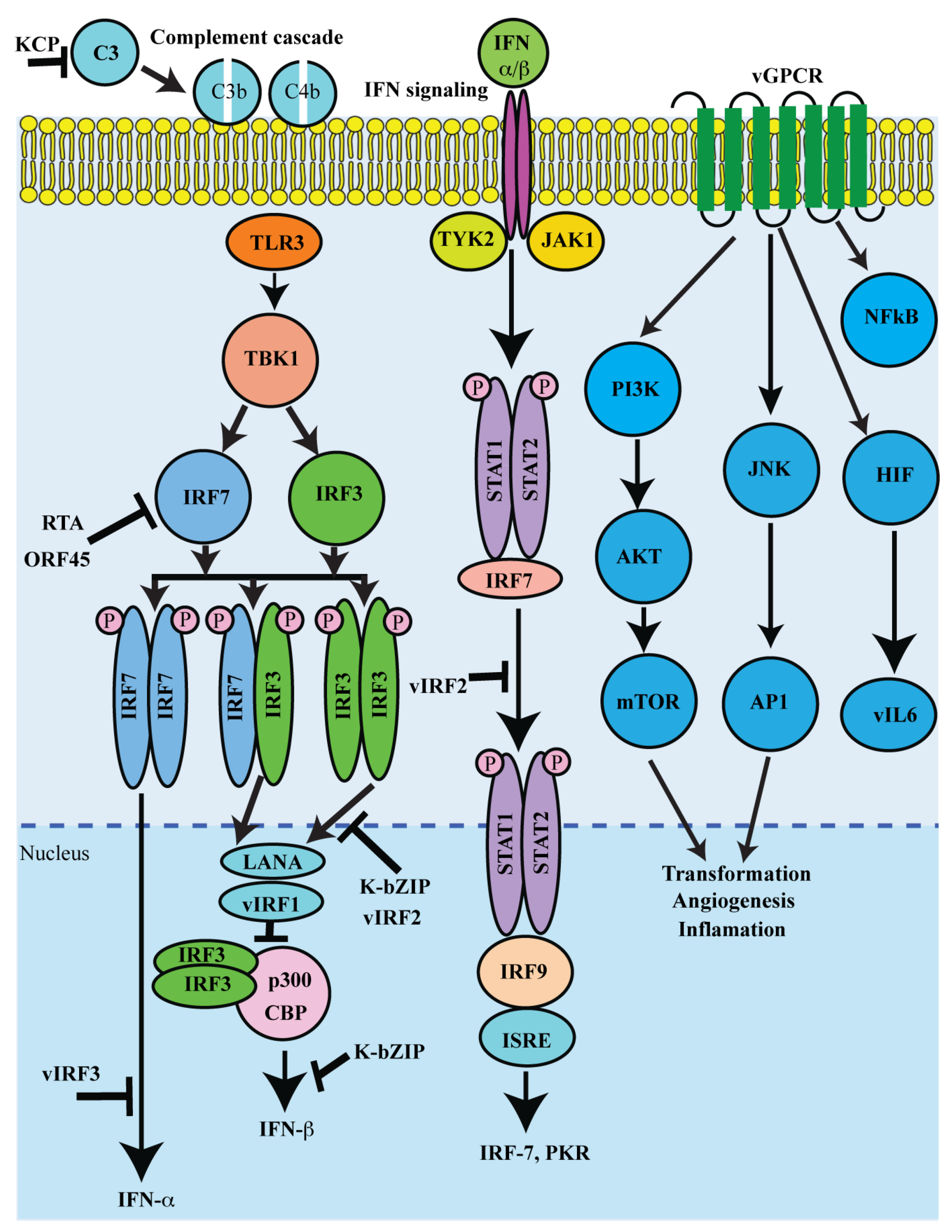
5. Lytic Proteins in Controlling Immune Regulation and Pathogenesis

{kind=link}
{kind=link}
{kind=link}
| KSHV genes | KSHV proteins | Function | References |
|---|---|---|---|
| K1 | Variable ITAM-Containing Protein (VIP) | Type I transmembrane signaling protein containing a functional immunoreceptor tyrosine-based activation motif. Regulate membrane transport in B cells. | [203] |
| K2 | Viral Interleukin-6 (vIL-6) | Homologues of cellular IL-6. Activate JAK/STAT, MAPK, and PI3K/Akt signaling pathways to regulate B-cell proliferation. | [51,204] |
| K3/K5 | Modulator of immune recognition (MIR1/MIR2) | Viral E3 ligases capable of ubiquitinating MHC-I, ICAM-1, B7-2, Tetherin (CD317/BST2), DC-SIGN, and DC-SIGNR. | [205,206] |
| K4/K4.1/K6 | Viral CC-Chemokine Ligands (vCCLs) | Homologues of cellular chemokines: viral CC-chemokine ligand 1 vCCL1 (vMIP1), vCCL2 (vMIP2), and vCCL3 (vMIP3), respectively. Blocks signaling through chemokine receptors. | [207,208] |
| K7 | Viral Inhibitor of Apoptosis (vIAP) | Interact with cellular proteins PLIC1, caspase 3/Bcl-2, CAML, Vps34, and promote cell survival during lytic replication. | [209,210] |
| K9/K10/K11 | KSHV interferon regulatory factors (vIRF-1, vIRF-2, vIRF-3 and vIRF-4) | Homologues of cellular interferon: Inhibitor of IFN1, p53, NFκB RelA, and p300. | [211,212] |
| K14 | vOX2 or vCD200 | Homologues of cellular OX2. A negative regulator of inflammatory signaling and surface glycoproteins. | [213,214] |
| K15 | Viral membrane protein | Regulation of cellular signaling to induce various pro-survival and paracrine-mediated pro- angiogenic cellular cytokines and chemokines, including IL6, IL8, IL-1a/b, CXCL3, and Cox2. | [215,216] |
| ORF4 | KSHV complement Control protein (KCP) | Homologue to cellular RCA. Regulate complement activation by increasing the decay of the classical C3 convertase. | [217,218,219] |
| ORF45 | ORF45 | Inhibit type1 IFN induction by sequestering the cellular interferon regulatory factor-7 to cytoplasm. | [220,221] |
| ORF63 | ORF63 | Homologue to cellular inflammasome complex NLRP1. | [222] |
| ORF64 | Viral deubiquitinase | A non specific deubiquitinase, shown to deubiquitinate RIG-I to suppress RIG-I-mediated activation of the IFNb. | [223] |
| ORF74 | Viral G-protein-coupled receptor (vGPCR) | Homologue of cellular IL-8 receptor. vGPCR induce secretion of proinflammatory cytokines and angiogenic growth factors. | [200,224] |
| ORF75 | ORF75 | A viral effector for the degradation of ND10 proteins. | [225,226] |
| PAN RNA | Polyadenylated Nuclear RNA | Modulator of viral gene expression. | [227,228,229,230] |

6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sturzl, M.; Zietz, C.; Monini, P.; Ensoli, B. Human herpesvirus-8 and Kaposi’s sarcoma: Relationship with the multistep concept of tumorigenesis. Adv. Cancer Res. 2001, 81, 125–159. [Google Scholar] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar]
- Uldrick, T.S.; Wang, V.; O'Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef]
- Dittmer, D.; Stoddart, C.; Renne, R.; Linquist-Stepps, V.; Moreno, M.E.; Bare, C.; McCune, J.M.; Ganem, D. Experimental transmission of Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 1999, 190, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 2014, 6, 4024–4046. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lagunoff, M. Establishment and maintenance of Kaposi’s sarcoma-associated herpesvirus latency in B cells. J. Virol. 2005, 79, 14383–14391. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Wang, S.E.; Wu, F.Y.; Yu, Y.; Hayward, G.S. CCAAT/enhancer-binding protein-α is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 2003, 77, 9590–9612. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Young, A.; Sun, R. Auto-activation of the rta gene of human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2000, 81, 3043–3048. [Google Scholar] [PubMed]
- Song, M.J.; Deng, H.; Sun, R. Comparative study of regulation of RTA-responsive genes in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 2003, 77, 9451–9462. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Q.; Maul, G.G.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Dual role of replication and transcription activator. J. Virol. 2006, 80, 12171–12186. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: Identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Xu, Y.; Cei, S.A.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 2002, 76, 7890–7896. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Sharma, N.; Murakami, M.; Robertson, E.S. Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: A novel mechanism for establishment of latency. J. Virol. 2005, 79, 7453–7465. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, F.; Ye, F.; Gao, S.J. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 2008, 379, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jκ, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ueda, K.; Sakakibara, S.; Okuno, T.; Parravicini, C.; Corbellino, M.; Yamanishi, K. Activation of latent Kaposi’s sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. USA 2001, 98, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed]
- Woodard, C.; Shamay, M.; Liao, G.; Zhu, J.; Ng, A.N.; Li, R.; Newman, R.; Rho, H.S.; Hu, J.; Wan, J.; et al. Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog. 2012, 8, e1002972. [Google Scholar] [CrossRef]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi’s sarcoma associated herpesvirus reactivation. BMC Genomics 2013, 14, 824. [Google Scholar] [CrossRef]
- Chang, P.C.; Izumiya, Y.; Wu, C.Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.J.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, B.G.; Verma, S.C.; Lan, K.; Cotter, M.A.; Woodman, Z.L.; Robertson, E.S. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 2006, 351, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Lim, C.; Lee, J.Y.; Song, Y.J.; Park, J.; Choe, J.; Seo, T. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 2010, 394, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Chang, P.C.; Huerta, S.; Izumiya, C.; Davis, R.; Tepper, C.G.; Kim, K.Y.; Shevchenko, B.; Wang, D.H.; Jung, J.U.; et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 2012, 287, 5806–5818. [Google Scholar]
- Campbell, M.; Izumiya, Y. Post-Translational Modifications of Kaposi’s Sarcoma-Associated Herpesvirus Regulatory Proteins—SUMO and KSHV. Front. Microbiol. 2012, 3, 31. [Google Scholar] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed]
- Stuber, G.; Mattsson, K.; Flaberg, E.; Kati, E.; Markasz, L.; Sheldon, J.A.; Klein, G.; Schulz, T.F.; Szekely, L. HHV-8 encoded LANA-1 alters the higher organization of the cell nucleus. Mol. Cancer 2007, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Huerta, S.B.; Izumiya, C.; Wang, D.H.; Martinez, A.; Shevchenko, B.; Kung, H.J.; Campbell, M.; Izumiya, Y. Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J. Virol. 2013, 87, 6782–6793. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Ye, F.; Bedolla, R.; Huang, Y.; Meng, J.; Qian, L.; Pan, H.; Zhou, F.; Moody, R.; Wagner, B.; et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J. Clin. Investig. 2012, 122, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; O’Hearn, P.; Kimball, L.; Chandran, B.; Corey, L. Activation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 2001, 75, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xue, M.; Qin, D.; Zhu, X.; Wang, C.; Zhu, J.; Hao, T.; Cheng, L.; Chen, X.; Bai, Z.; et al. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3beta signaling pathway. PLoS One 2013, 8, e53145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guo, Y.; Yao, S.; Yan, Q.; Xue, M.; Hao, T.; Zhou, F.; Zhu, J.; Qin, D.; Lu, C. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: Role of the AKT signaling pathway. Oncogene 2014, 33, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Roupelieva, M.; Griffiths, S.J.; Kremmer, E.; Meisterernst, M.; Viejo-Borbolla, A.; Schulz, T.; Haas, J. Kaposi’s sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J. Gen. Virol. 2010, 91, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Qin, D.; Lv, Z.; Zhu, X.; Ma, X.; Yan, Q.; Zeng, Y.; Guo, Y.; Feng, N.; Lu, C. Herpes simplex virus type 2 triggers reactivation of Kaposi’s sarcoma-associated herpesvirus from latency and collaborates with HIV-1 Tat. PLoS One 2012, 7, e31652. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zeng, Y.; Huang, Z.; Huang, L.; Qian, C.; Tang, G.; Qin, D. Human herpesvirus 6 activates lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus. Am. J. Pathol. 2005, 166, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A. Skin diseases associated with human herpesvirus 6, 7, and 8 infection. J. Investig. Dermatol. Symp. Proc. 2001, 6, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Rady, P.L.; Yen, A.; Rollefson, J.L.; Orengo, I.; Bruce, S.; Hughes, T.K.; Tyring, S.K. Herpesvirus-like DNA sequences in non-Kaposi’s sarcoma skin lesions of transplant patients. Lancet 1995, 345, 1339–1340. [Google Scholar] [CrossRef] [PubMed]
- Mercader, M.; Taddeo, B.; Panella, J.R.; Chandran, B.; Nickoloff, B.J.; Foreman, K.E. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am. J. Pathol. 2000, 156, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1α to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Cai, S.; Zhu, C.; Verma, S.C.; Choi, J.Y.; Robertson, E.S. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. 2013, 9, e1003750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 enhances hypoxia-induced Kaposi’s sarcoma-associated herpesvirus reactivation through RBP-Jκ. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Ruffels, J.; Griffin, M.; Dickenson, J.M. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 2004, 483, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; DeCotiis, J.; Giron, M.; Palmeri, D.; Lukac, D.M. Histone deacetylase classes I and II regulate Kaposi’s sarcoma-associated herpesvirus reactivation. J. Virol. 2014, 88, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, M.; Zhou, F.; Ye, F.; Gao, S.J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 2014, 88, 6355–6367. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Walker, L.R.; Whitehouse, A.; Cook, P.P.; Akula, S.M. Resveratrol inhibits KSHV reactivation by lowering the levels of cellular EGR-1. PLoS One 2012, 7, e33364. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papouskova, I.; Tarrant, M.; Pari, G.S. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Gradoville, L.; Gerlach, J.; Grogan, E.; Shedd, D.; Nikiforow, S.; Metroka, C.; Miller, G. Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 2000, 74, 6207–6212. [Google Scholar] [CrossRef] [PubMed]
- Cousins, E.; Nicholas, J. Molecular biology of human herpesvirus 8: Novel functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 2014, 193, 227–268. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; AuCoin, D.P.; Huete, A.R.; Cei, S.A.; Hanson, L.J.; Pari, G.S. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 2005, 79, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for γ2-herpesvirus lytic replication. Mol. Cell Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, S.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Palmeri, D.; Krishnan, R.; Marin, R.; Aris, V.M.; Soteropoulos, P.; Lukac, D.M. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 2008, 82, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Song, M.J.; Chu, J.T.; Sun, R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J. Virol. 2002, 76, 8252–8264. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Brown, H.J.; Wu, T.T.; Sun, R. Transcription activation of polyadenylated nuclear rna by rta in human herpesvirus 8/Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.; Gwack, Y.; Hwang, S.; Choe, J. Kaposi’s sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cells 2002, 14, 185–191. [Google Scholar] [PubMed]
- McDowell, M.E.; Purushothaman, P.; Rossetto, C.C.; Pari, G.S.; Verma, S.C. Phosphorylation of Kaposi’s sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J. Virol. 2013, 87, 8038–8052. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Susilarini, N.K.; Pari, G.S. Interaction of Kaposi’s sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol. 2011, 85, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Ishikawa, K.; Nishimura, K.; Sakakibara, S.; Do, E.; Yamanishi, K. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 2002, 76, 12044–12054. [Google Scholar] [CrossRef] [PubMed]
- Bowser, B.S.; Morris, S.; Song, M.J.; Sun, R.; Damania, B. Characterization of Kaposi’s sarcoma-associated herpesvirus (KSHV) K1 promoter activation by Rta. Virology 2006, 348, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus small capsid protein by bacterial artificial chromosome-based mutagenesis. Virology 2010, 407, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Zheng, Z.M. Gene structure and expression of Kaposi’s sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 2006, 80, 11968–11981. [Google Scholar] [CrossRef] [PubMed]
- Kronstad, L.M.; Brulois, K.F.; Jung, J.U.; Glaunsinger, B.A. Reinitiation after translation of two upstream open reading frames (ORF) governs expression of the ORF35–37 Kaposi’s sarcoma-associated herpesvirus polycistronic mRNA. J. Virol. 2014, 88, 6512–6518. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Means, R.E.; Choi, J.K.; Lee, B.S.; Jung, J.U. Kaposi's sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 2002, 76, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Seaman, W.T.; Quinlivan, E.B. Lytic switch protein (ORF50) response element in the Kaposi’s sarcoma-associated herpesvirus K8 promoter is located within but does not require a palindromic structure. Virology 2003, 310, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: A potential mechanism for virus-mediated control of latency. J. Virol. 2004, 78, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, J.; Grundhoff, A.; Ganem, D. Exploring the DNA binding interactions of the Kaposi’s sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J. Virol. 2006, 80, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, F.; Xie, J.; Kuhne, K.; Gao, S.J. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 2009, 386, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, J.; Lynch, S.J.; Lukac, D.M.; Ganem, D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev. 2002, 16, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Tsao, E.H.; Webb, B.L.; Ye, H.; Dalton-Griffin, L.; Tsantoulas, C.; Gale, C.V.; Du, M.Q.; Whitehouse, A.; Kellam, P. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 2007, 81, 13578–13586. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shedd, D.; Miller, G. An Sp1 response element in the Kaposi’s sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 2005, 79, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Khadim, F.; Spadavecchia, S.; Palmeri, D.; Lukac, D.M. Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 2007, 81, 8451–8467. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Shedd, D.; Miller, G. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 2005, 79, 8750–8763. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Boonsiri, J.; Wang, S.S.; Chen, L.Y.; Miller, G. Binding of RBP-Jκ (CSL) protein to the promoter of the Kaposi’s sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 2010, 398, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.M.; Wilson, A.C. Wide-scale use of Notch signaling factor CSL/RBP-Jκ in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J. Virol. 2010, 84, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Saha, A.; Dzeng, R.K.; Robertson, E.S. The RBP-Jκ binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 2012, 8, e1002479. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Choudhuri, T.; Kuppers, D.A.; Robertson, E.S. Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 2006, 351, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Dittmer, D.P.; Shin, Y.C.; Hong, Y.; Jung, J.U. Role of Notch signal transduction in Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 2005, 79, 14371–14382. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Baek, H.J.; Nakamura, H.; Lee, S.H.; Meisterernst, M.; Roeder, R.G.; Jung, J.U. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell Biol. 2003, 23, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cai, Q.; Lu, J.; Jha, H.C.; Robertson, E.S. Upregulation of cellular Bcl-2 by the KSHV encoded RTA promotes virion production. PLoS One 2011, 6, e23892. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S.; Wu, M.H.; Geng, Y.; Wood, C. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi’s sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 2001, 75, 11961–11973. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wood, C. The transcriptional repressor K-RBP modulates RTA-mediated transactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6294–6306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wen, H.J.; Minhas, V.; Wood, C. The zinc finger DNA-binding domain of K-RBP plays an important role in regulating Kaposi’s sarcoma-associated herpesvirus RTA-mediated gene expression. Virology 2009, 391, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.J. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Tang, Y.; Lin, S.F.; Kung, H.J.; Giam, C.Z. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 2003, 77, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Gao, Y.; Yamboliev, I.; Papouskova, I.; Pari, G. Transcriptional repression of K-Rta by Kaposi’s sarcoma-associated herpesvirus K-bZIP is not required for oriLyt-dependent DNA replication. Virology 2007, 369, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Fujimuro, M.; Zong, J.; Hayward, S.D.; Hayward, G.S. Role of CCAAT/enhancer-binding protein α (C/EBPα) in activation of the Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 2003, 77, 600–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, Y. Essential role of RBP-Jκ in activation of the K8 delayed-early promoter of Kaposi’s sarcoma-associated herpesvirus by ORF50/RTA. Virology 2007, 359, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Liang, D.; Gao, Y.; Xia, T.; Robertson, E.S.; Lan, K. Kaposi’s sarcoma-associated herpesvirus RTA activates the processivity factor ORF59 through interaction with RBP-Jκ and a cis-acting RTA responsive element. Virology 2008, 380, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Wang, S.S.; Chen, L.Y.; Hung, C.H.; Huang, H.Y.; Shih, Y.J.; Yen, J.B.; Liou, J.Y.; Chen, L.W. ORF50-dependent and ORF50-independent activation of the ORF45 gene of Kaposi’s sarcoma-associated herpesvirus. Virology 2013, 442, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Gavina, A.; Palmeri, D.; Lukac, D.M. The cellular peptidyl-prolyl cis/trans isomerase Pin1 regulates reactivation of Kaposi’s sarcoma-associated herpesvirus from latency. J. Virol. 2014, 88, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Yuan, Y. A virally encoded small peptide regulates RTA stability and facilitates Kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2013, 87, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Chan, M.Y.; Zhu, F.X.; Lukac, D.M.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 2004, 78, 8615–8629. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s sarcoma-associated herpesvirus K-Rta exhibits SUMO-targeting ubiquitin ligase (STUbL) like activity and is essential for viral reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Swaminathan, S. Kaposi’s sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol. 2006, 80, 5251–5260. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Pripuzova, N.; McCoy, J.P.; Gao, S.J.; Zheng, Z.M. Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8α, and K8.1 and the production of infectious virus. J. Virol. 2007, 81, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Spadavecchia, S.; Carroll, K.D.; Lukac, D.M. Promoter- and cell-specific transcriptional transactivation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 2007, 81, 13299–13314. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Blackbourn, D.J.; Cheng, M.F.; Hayward, G.S.; Clements, J.B. Functional co-operation between the Kaposi’s sarcoma-associated herpesvirus ORF57 and ORF50 regulatory proteins. J. Gen. Virol. 2004, 85, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Hunter, O.V.; Sei, E.; Richardson, R.B.; Conrad, N.K. Chromatin immunoprecipitation and microarray analysis suggest functional cooperation between Kaposi’s Sarcoma-associated herpesvirus ORF57 and K-bZIP. J. Virol. 2013, 87, 4005–4016. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Blackbourn, D.J.; Clements, J.B. The evolutionarily conserved Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J. Biol. Chem. 2004, 279, 33001–33011. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 in viral RNA processing. Front. Biosci. (Landmark Ed) 2009, 14, 1516–1528. [Google Scholar] [CrossRef]
- Nekorchuk, M.; Han, Z.; Hsieh, T.T.; Swaminathan, S. Kaposi’s sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J. Virol. 2007, 81, 9990–9998. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Kang, J.G.; Majerciak, V.; Le, S.Y.; Liewehr, D.J.; Steinberg, S.M.; Zheng, Z.M. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5' end to interact with viral ORF57 and cellular PABPC1. Int. J. Biol. Sci. 2011, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Majerciak, V.; Kruhlak, M.; Zheng, Z.M. Interplay between polyadenylate-binding protein 1 and Kaposi’s sarcoma-associated herpesvirus ORF57 in accumulation of polyadenylated nuclear RNA, a viral long noncoding RNA. J. Virol. 2013, 87, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.B.; Patel, D.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010, 6, e1000799. [Google Scholar] [CrossRef]
- Majerciak, V.; Uranishi, H.; Kruhlak, M.; Pilkington, G.R.; Massimelli, M.J.; Bear, J.; Pavlakis, G.N.; Felber, B.K.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J. Virol. 2011, 85, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Robinson, D.R.; Miller, G.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 1999, 73, 1909–1917. [Google Scholar] [PubMed]
- Tang, S.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus K8 exon 3 contains three 5'-splice sites and harbors a K8.1 transcription start site. J. Biol. Chem. 2002, 277, 14547–14556. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.; Chen, L.Y.; Izumiya, C.; Luciw, P.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: Physical association and promoter-dependent transcriptional repression. J. Virol. 2003, 77, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.J. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Kato-Noah, T.; Xu, Y.; Rossetto, C.C.; Colletti, K.; Papouskova, I.; Pari, G.S. Overexpression of the kaposi’s sarcoma-associated herpesvirus transactivator K-Rta can complement a K-bZIP deletion BACmid and yields an enhanced growth phenotype. J. Virol. 2007, 81, 13519–13532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sathish, N.; Hollow, C.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus open reading frame K8 by bacterial artificial chromosome-based mutagenesis. J. Virol. 2011, 85, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Flamand, L. Kaposi’s sarcoma-associated herpesvirus K-bZIP protein is necessary for lytic viral gene expression, DNA replication, and virion production in primary effusion lymphoma cell lines. J. Virol. 2009, 83, 5869–5880. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Wang, S.E.; Tang, Q.Q.; Fujimuro, M.; Chiou, C.J.; Zheng, Q.; Chen, H.; Hayward, S.D.; Lane, M.D.; Hayward, G.S. Cell cycle arrest by Kaposi’s sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein α and p21(CIP-1). J. Virol. 2003, 77, 8893–8914. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Yamboliev, I.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 K-bZIP modulates latency-associated nuclear protein-mediated suppression of lytic origin-dependent DNA synthesis. J. Virol. 2009, 83, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Ellison, T.J.; Yeh, E.T.; Jung, J.U.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 2005, 79, 9912–9925. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Soucy-Faulkner, A.; Grandvaux, N.; Flamand, L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 2007, 81, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Gravel, A.; Flamand, L. Repression of interferon-α stimulated genes expression by Kaposi’s sarcoma-associated herpesvirus K-bZIP protein. Virology 2010, 408, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.P.; Tang, Q. Leucine zipper domain is required for Kaposi sarcoma-associated herpesvirus (KSHV) K-bZIP protein to interact with histone deacetylase and is important for KSHV replication. J. Biol. Chem. 2012, 287, 15622–15634. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; van Geelen, A.; Wang, D.H.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 2007, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. bZIP proteins of human γ-herpesviruses. J. Gen. Virol. 2003, 84, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ciustea, M.; Ricciardi, R.P. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 2005, 340, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of Kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.; Zong, J.; Ciufo, D.; Guo, H.G.; Reitz, M.S.; Hayward, G.S. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J. Virol. 1997, 71, 1963–1974. [Google Scholar] [PubMed]
- Peng, C.; Chen, J.; Tang, W.; Liu, C.; Chen, X. Kaposi’s sarcoma-associated herpesvirus ORF6 gene is essential in viral lytic replication. PLoS One 2014, 9, e99542. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Cohen, A.; Kazimirsky, G.; Dovrat, S.; Rubinfeld, H.; Brodie, C.; Sarid, R. Role of protein kinase C delta in reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2004, 78, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.W.; Bryan, B.A.; Dyson, O.F.; Weidner, D.A.; Chintalgattu, V.; Akula, S.M. Raf/MEK/ERK signalling triggers reactivation of Kaposi’s sarcoma-associated herpesvirus latency. J. Gen. Virol. 2006, 87, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Choudhuri, T.; Murakami, M.; Kuppers, D.A.; Robertson, E.S. Intracellular activated Notch1 is critical for proliferation of Kaposi’s sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 2006, 80, 6411–6419. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Weidner-Glunde, M.; Varjosalo, M.; Rainio, E.M.; Lehtonen, A.; Schulz, T.F.; Koskinen, P.J.; Taipale, J.; Ojala, P.M. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Feng, N.; Fan, W.; Ma, X.; Yan, Q.; Lv, Z.; Zeng, Y.; Zhu, J.; Lu, C. Activation of PI3K/AKT and ERK MAPK signal pathways is required for the induction of lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus by herpes simplex virus type 1. BMC Microbiol. 2011, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Verma, D.; Mosbruger, T.; Swaminathan, S. CTCF and Rad21 act as host cell restriction factors for Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014, 10, e1003880. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Doganay, S.; Chung, B.; Toth, Z.; Brulois, K.; Lee, S.; Kanketayeva, Z.; Feng, P.; Ha, T.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) targets expression of cellular IRF4 and the Myc gene to facilitate lytic replication. J. Virol. 2014, 88, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Yang, L.; Mancl, M.E.; Barnes, B.J. Modulation of interferon regulatory factor 5 activities by the Kaposi sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3 contributes to immune evasion and lytic induction. J. Interferon Cytokine Res. 2011, 31, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Fu, B.; Liang, Q.; Myoung, J.; Zhu, F. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem. 2011, 286, 41171–41182. [Google Scholar] [CrossRef] [PubMed]
- Zoeteweij, J.P.; Moses, A.V.; Rinderknecht, A.S.; Davis, D.A.; Overwijk, W.W.; Yarchoan, R.; Orenstein, J.M.; Blauvelt, A. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Krishnan, H.H.; Naranatt, P.P.; Zeng, L.; Smith, M.S.; Chandran, B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 2005, 79, 10308–10329. [Google Scholar] [CrossRef] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 2: Pathogenesis, Castleman’s disease, and pleural effusion lymphoma. Lancet Infect. Dis. 2002, 2, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.A.; Bala, K.; Busche, G.; Weidner-Glunde, M.; Santag, S.; Kati, S.; Gramolelli, S.; Damas, M.; Dittrich-Breiholz, O.; Kracht, M.; et al. The inflammatory kinase MAP4K4 promotes reactivation of Kaposi’s sarcoma herpesvirus and enhances the invasiveness of infected endothelial cells. PLoS Pathog. 2013, 9, e1003737. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Chen, H.; Shamay, M.; Zheng, Q.; Hayward, G.S. Early activation of the Kaposi’s sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 2004, 78, 4248–4267. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Pan, H.; Yoo, S.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 2005, 79, 15027–15037. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Brodie, C.; Sarid, R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2006, 87, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Bu, W.; Palmeri, D.; Spadavecchia, S.; Lynch, S.J.; Marras, S.A.; Tyagi, S.; Lukac, D.M. Kaposi’s Sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 2006, 80, 9697–9709. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Murakami, M.; Si, H.; Robertson, E.S. A potential α-helix motif in the amino terminus of LANA encoded by Kaposi’s sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1α in normoxia. J. Virol. 2007, 81, 10413–10423. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Kenerson, H.L.; Yeung, R.S.; Lagunoff, M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 2006, 80, 10802–10812. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Joo, C.H.; Gack, M.U.; Lee, H.R.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1α to induce vascular endothelial growth factor expression. Cancer Res. 2008, 68, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Damania, B. KSHV and the toll of innate immune activation. Cell Cycle 2009, 8, 3246–3247. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shahir, A.M.; Sha, J.; Feng, Z.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.A.; Punjabi, A.S.; Lagunoff, M. Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J. Virol. 2008, 82, 8771–8779. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, A.S.; Carroll, P.A.; Chen, L.; Lagunoff, M. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 2007, 81, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Hartmann, T.; Burger, J.A.; Schraufstatter, I. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene 2005, 24, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Okabe, K.; Fujimuro, M.; Sugiyama, K.; Yokosawa, H.; Seya, T.; Matsuda, T. Physical and functional interactions between STAT3 and Kaposi’s sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 2006, 580, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Choi, J.Y.; Ma, M.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus inhibits interleukin-4-mediated STAT6 phosphorylation to regulate apoptosis and maintain latency. J. Virol. 2010, 84, 11134–11144. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Xie, J.P.; Kang, T.; Greene, W.; Kuhne, K.; Lei, X.F.; Li, Q.H.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-κB-mediated suppression of the AP-1 pathway: A novel mechanism of virus control of latency. J. Virol. 2008, 82, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; Hsia, D.; Ellison, T.J.; Luciw, P.A.; Kung, H.J. NF-κB serves as a cellular sensor of Kaposi’s sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jκ coactivator. J. Virol. 2009, 83, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; Song, M.J.; Deng, H.; Wu, T.T.; Cheng, G.; Sun, R. NF-κB inhibits γ-herpesvirus lytic replication. J. Virol. 2003, 77, 8532–8540. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Ganem, D. Effects of NF-κB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 2008, 375, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.J.; Deng, J.H.; Zhou, F.C. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 2003, 77, 9738–9749. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, S.; Sharma-Walia, N.; Veettil, M.V.; Raghu, H.; Sivakumar, R.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 2007, 81, 3949–3968. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; et al. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances γ2-herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 2012, 3, 401. [Google Scholar] [PubMed]
- Lagunoff, M.; Majeti, R.; Weiss, A.; Ganem, D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1999, 96, 5704–5709. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.C.; Damania, B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Galisteo, R.; Molinolo, A.A.; Wetzker, R.; Hirsch, E.; Gutkind, J.S. PI3Kγ mediates kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 2011, 19, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.; Chaisuparat, R.; Hu, J.; Ramsdell, A.K.; Manning, B.D.; Sausville, E.A.; Sawai, E.T.; Molinolo, A.; Gutkind, J.S.; Montaner, S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 2006, 10, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Kuang, E.; Li, W.; Avey, D.; Li, X.; Turpin, Z.; Valdes, A.; Brulois, K.; Myoung, J.; Zhu, F. Activation of p90 Ribosomal S6 Kinases (RSKs) by ORF45 of Kaposi Sarcoma-Associated Herpesvirus is Critical for Optimal Production of Infectious Viruses. J. Virol. 2015, 89, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Maeng, H.; Young, D.P.; Prakash, O.; Fayad, L.E.; Younes, A.; Samaniego, F. K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood 2007, 109, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Teoh, G.; Raje, N.; Treon, S.P.; Tai, Y.T.; Shima, Y.; Anderson, K.C. Characterization of signaling cascades triggered by human interleukin-6 versus Kaposi’s sarcoma-associated herpes virus-encoded viral interleukin 6. Clin. Cancer Res. 2000, 6, 1180–1189. [Google Scholar] [PubMed]
- Wang, Q.J.; Jenkins, F.J.; Jacobson, L.P.; Meng, Y.X.; Pellett, P.E.; Kingsley, L.A.; Kousoulas, K.G.; Baghian, A.; Rinaldo, C.R., Jr. CD8+ cytotoxic T lymphocyte responses to lytic proteins of human herpes virus 8 in human immunodeficiency virus type 1-infected and -uninfected individuals. J. Infect. Dis. 2000, 182, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Bynoe, M.O.; Karki, R.; Tartell, M.A.; Means, R.E. Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins down regulate both DC-SIGN and DC-SIGNR. PLoS One 2013, 8, e58056. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.S.; Grone, H.J.; Rocken, M.; Klier, C.; Gu, S.; Wank, R.; Proudfoot, A.E.; Nelson, P.J.; Weber, C. Selective recruitment of Th2-type cells and evasion from a cytotoxic immune response mediated by viral macrophage inhibitory protein-II. Eur. J. Immunol. 2001, 31, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Kledal, T.N.; Rosenkilde, M.M.; Coulin, F.; Simmons, G.; Johnsen, A.H.; Alouani, S.; Power, C.A.; Luttichau, H.R.; Gerstoft, J.; Clapham, P.R.; et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 1997, 277, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Dong, X.; Negaard, A.; Feng, P. Kaposi’s sarcoma-associated herpesvirus K7 induces viral G protein-coupled receptor degradation and reduces its tumorigenicity. PLoS Pathog. 2008, 4, e1000157. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Sharp, T.V.; Koumi, A.; Koentges, G.; Boshoff, C. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 2002, 21, 2602–2615. [Google Scholar] [CrossRef] [PubMed]
- Burysek, L.; Yeow, W.S.; Lubyova, B.; Kellum, M.; Schafer, S.L.; Huang, Y.Q.; Pitha, P.M. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999, 73, 7334–7342. [Google Scholar] [PubMed]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Foster-Cuevas, M.; Wright, G.J.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 2004, 78, 7667–7676. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, S.A.; Gracie, J.A.; McInnes, I.B.; Blackbourn, D.J. Inhibition of neutrophil function by the Kaposi’s sarcoma-associated herpesvirus vOX2 protein. AIDS 2005, 19, 1907–1910. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Pietrek, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Modulation of host gene expression by the K15 protein of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brinkmann, M.M.; Pietrek, M.; Ottinger, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Functional characterization of the M-type K15-encoded membrane protein of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2007, 88, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Mullick, J.; Bernet, J.; Singh, A.K.; Lambris, J.D.; Sahu, A. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) open reading frame 4 protein (kaposica) is a functional homolog of complement control proteins. J. Virol. 2003, 77, 3878–3881. [Google Scholar] [CrossRef] [PubMed]
- Mullick, J.; Singh, A.K.; Panse, Y.; Yadav, V.; Bernet, J.; Sahu, A. Identification of functional domains in kaposica, the complement control protein homolog of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J. Virol. 2005, 79, 5850–5856. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Robinson, M.; O'Donnell, E.; Milligan, S.; Morgan, B.P.; Davison, A.J.; Blackbourn, D.J. Complement regulation by Kaposi’s sarcoma-associated herpesvirus ORF4 protein. J. Virol. 2003, 77, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 2002, 99, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Zhu, F.X.; Golub, E.E.; Liang, Q.; Yuan, Y. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi’s sarcoma-associated herpesvirus protein ORF45. J. Biol. Chem. 2011, 286, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Damania, B. Inhibition of the inflammasome response by a viral protein that interacts with NLRs. Commun. Integr. Biol. 2011, 4, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Nador, R.G.; Bai, F.; Bohenzky, R.A.; Russo, J.J.; Moore, P.S.; Chang, Y.; Knowles, D.M. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996, 70, 8218–8223. [Google Scholar] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Darricarrere, N.; Darnell, A.; Myoung, J.; Steitz, J.A. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Nichols, L.A.; Hassman, L.M.; Kedes, D.H.; Steitz, J.A. Tracking expression and subcellular localization of RNA and protein species using high-throughput single cell imaging flow cytometry. RNA 2012, 18, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J. Virol. 2011, 85, 13290–13297. [Google Scholar] [CrossRef] [PubMed]
- Monini, P.; Carlini, F.; Sturzl, M.; Rimessi, P.; Superti, F.; Franco, M.; Melucci-Vigo, G.; Cafaro, A.; Goletti, D.; Sgadari, C.; et al. α-interferon inhibits human herpesvirus 8 (HHV-8) reactivation in primary effusion lymphoma cells and reduces HHV-8 load in cultured peripheral blood mononuclear cells. J. Virol. 1999, 73, 4029–4041. [Google Scholar] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Gregory, S.M.; West, J.A.; Wollish, A.C.; Bennett, C.L.; Blackbourn, D.J.; Heise, M.T.; Damania, B. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 2013, 87, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Choi, W.C.; Lee, S.; Hwang, J.; Hwang, E.; Guchhait, K.; Haas, J.; Toth, Z.; Jeon, Y.H.; Oh, T.K.; et al. Jung Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat. Struct. Mol. Biol. 2011, 18, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Molden, J.; Chang, Y.; You, Y.; Moore, P.S.; Goldsmith, M.A. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 1997, 272, 19625–19631. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: Emerging insights into the molecular oncogenesis and treatment of Kaposi’s sarcoma. Cell Cycle 2007, 6, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Itami, M. Seropositivity of human herpesvirus-8 in patients with uveitis. Ocul. Immunol. Inflamm. 2002, 10, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, S.H.; Feng, P.; Chang, H.; Cho, N.H.; Jung, J.U. Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J. Virol. 2005, 79, 12173–12184. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Swamy, O.R.; Peng, X.; Tang, Z.Y.; Li, L.; Larson, J.E.; Cohen, J.C.; Gill, J.; Farr, G.; Wang, S.; et al. Activation of Src kinase Lyn by the Kaposi sarcoma-associated herpesvirus K1 protein: Implications for lymphomagenesis. Blood 2005, 105, 3987–3994. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Park, J.; Lee, B.S.; Lee, S.H.; Bram, R.J.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 2002, 76, 11491–11504. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Jung, J.U. Interplay between Kaposi’s sarcoma-associated herpesvirus and the innate immune system. Cytokine Growth Factor Rev. 2014, 25, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7–2 and modulates T cell costimulation. J. Clin. Investig. 2001, 107, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Albrecht, J.C.; Ensser, A.; Huang, Y.Q.; Li, J.J.; Friedman-Kien, A.E.; Fleckenstein, B. Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 1997, 71, 839–842. [Google Scholar] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Okruzhnov, Y.; Li, H.; Nicholas, J. Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 2001, 75, 10933–10940. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.T.; Wood, C.; Hill, M.; Epp, A.; Raport, C.J.; Schweickart, V.L.; Endo, Y.; Sasaki, T.; Simmons, G.; Boshoff, C.; et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 2000, 95, 1151–1157. [Google Scholar] [PubMed]
- Shiratori, I.; Yamaguchi, M.; Suzukawa, M.; Yamamoto, K.; Lanier, L.L.; Saito, T.; Arase, H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 2005, 175, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Proctor, D.; Blackbourn, D.J.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein mimics human molecular mechanisms for inhibition of the complement system. J. Biol. Chem. 2004, 279, 45093–45101. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Blackbourn, D.J.; Mark, L.; Proctor, D.G.; Blom, A.M. Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem. 2003, 278, 9283–9289. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Mark, L.; Blue, C.E.; Proctor, D.G.; Aitken, J.A.; Blom, A.M.; Blackbourn, D.J. Dissecting the regions of virion-associated Kaposi’s sarcoma-associated herpesvirus complement control protein required for complement regulation and cell binding. J. Virol. 2006, 80, 4068–4078. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Proctor, D.G.; Blackbourn, D.J.; Blom, A.M.; Spiller, O.B. Separation of decay-accelerating and cofactor functional activities of Kaposi’s sarcoma-associated herpesvirus complement control protein using monoclonal antibodies. Immunology 2008, 123, 228–238. [Google Scholar] [PubMed]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [PubMed]
- Sathish, N.; Zhu, F.X.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog. 2009, 5, e1000332. [Google Scholar] [CrossRef] [PubMed]
- Brousset, P.; Meggetto, F.; Laharrague, P.; Attal, M.; Delsol, G. Kaposi’s sarcoma-associated herpesvirus (KSHV) in bone marrow biopsy from patients with multiple myeloma: PCR amplification of orf26 but not orf72 and orf75 sequences. Br. J. Haematol. 2000, 108, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-κB activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Rozen, R.; Sathish, N.; Li, Y.; Yuan, Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4742–4750. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Kim, K.Y.; Chang, P.C.; Huerta, S.; Shevchenko, B.; Wang, D.H.; Izumiya, C.; Kung, H.J.; Izumiya, Y. A lytic viral long noncoding RNA modulates the function of a latent protein. J. Virol. 2014, 88, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular Biology of KSHV Lytic Reactivation. Viruses 2015, 7, 116-153. https://doi.org/10.3390/v7010116
Purushothaman P, Uppal T, Verma SC. Molecular Biology of KSHV Lytic Reactivation. Viruses. 2015; 7(1):116-153. https://doi.org/10.3390/v7010116
Chicago/Turabian StylePurushothaman, Pravinkumar, Timsy Uppal, and Subhash C. Verma. 2015. "Molecular Biology of KSHV Lytic Reactivation" Viruses 7, no. 1: 116-153. https://doi.org/10.3390/v7010116
APA StylePurushothaman, P., Uppal, T., & Verma, S. C. (2015). Molecular Biology of KSHV Lytic Reactivation. Viruses, 7(1), 116-153. https://doi.org/10.3390/v7010116