
KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control

Abstract
:1. Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation: A Primer
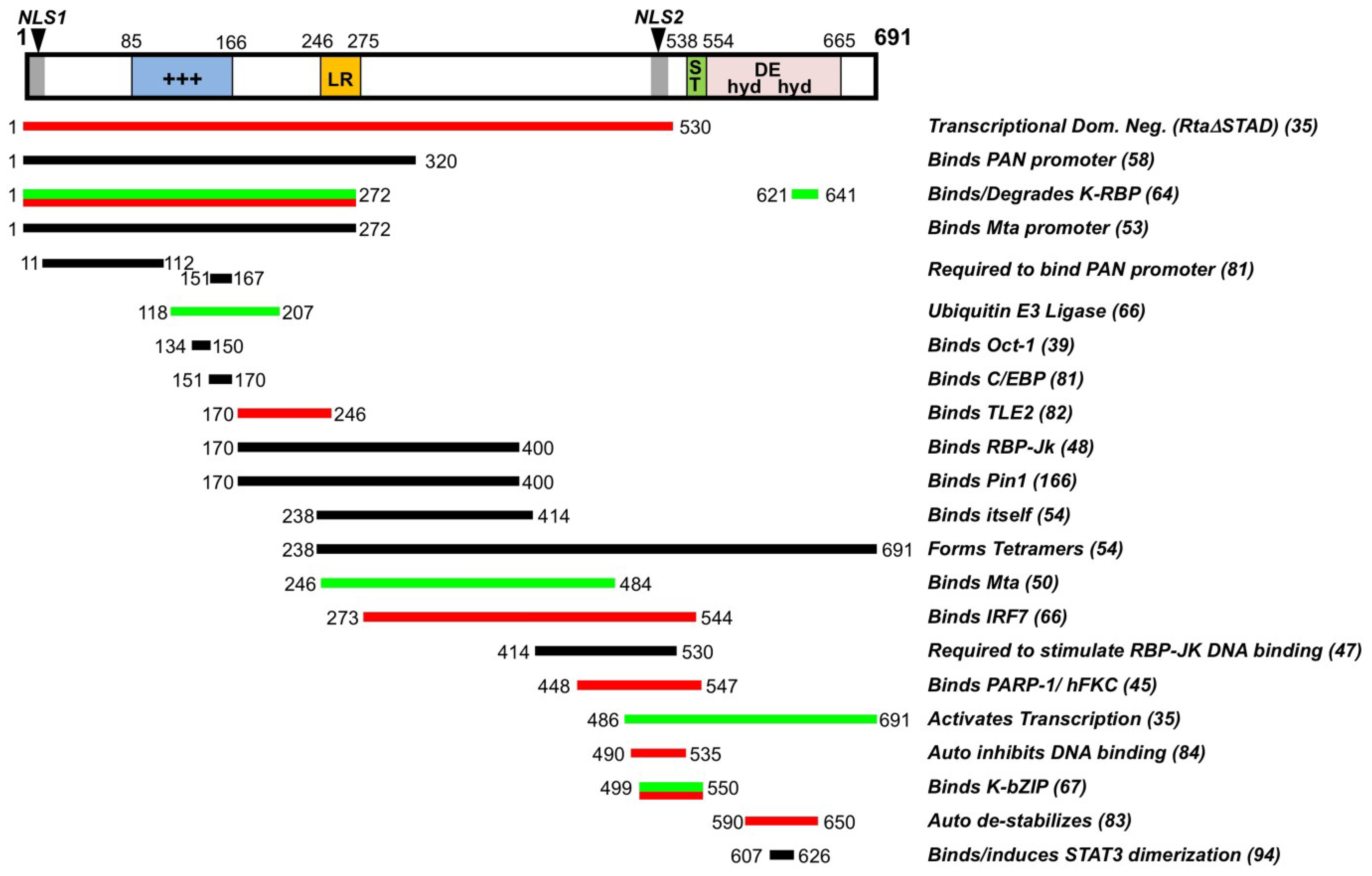
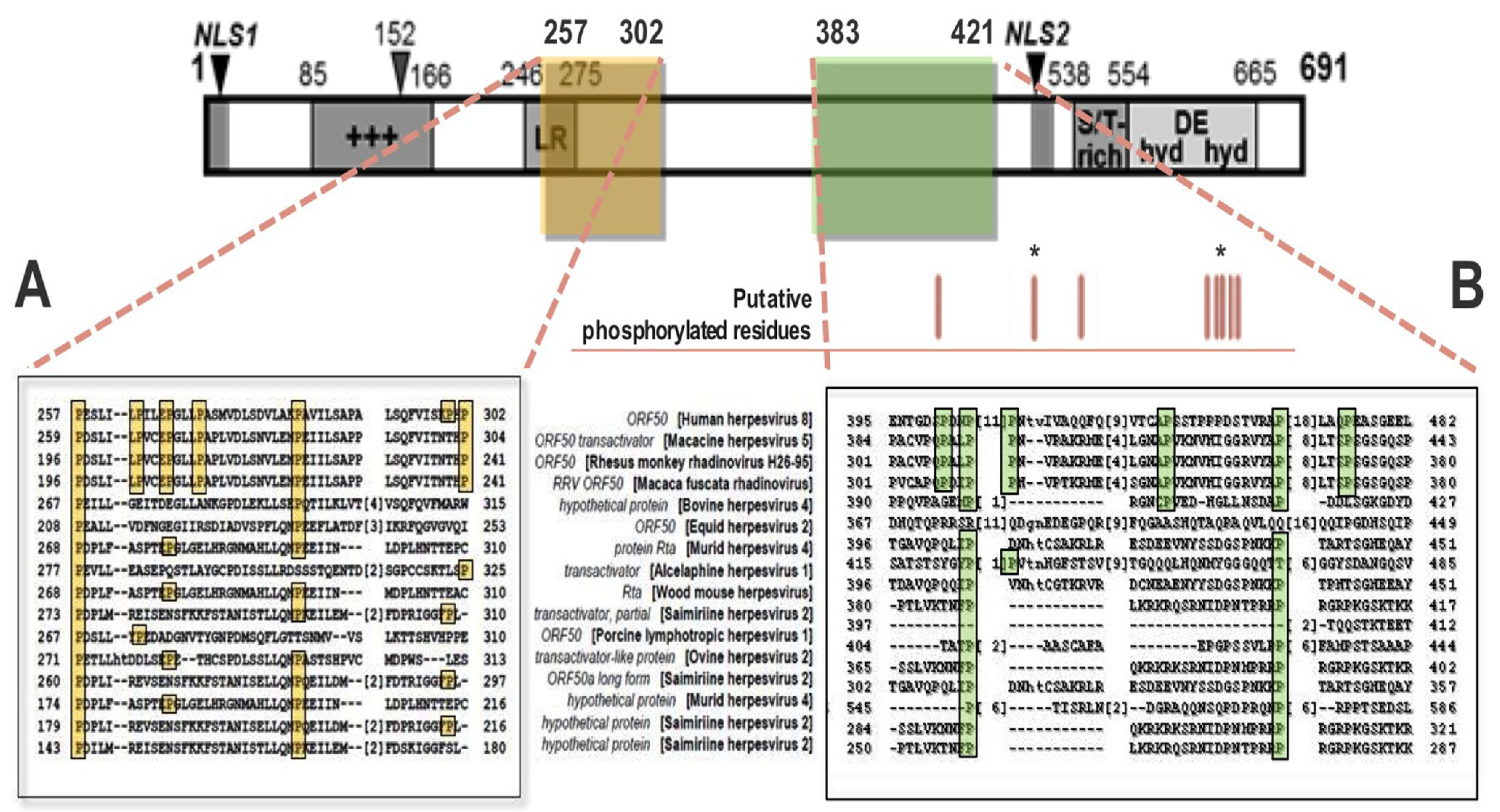
2. Function and Regulation of Rta Lytic Switch Protein

2.1. Mechanisms of Rta-Mediated Transactivation
2.2. Rta Positively and Negatively Interacts with Host and Viral Cofactors
2.3. RBP-Jk Is Essential for Rta-Mediated Transactivation
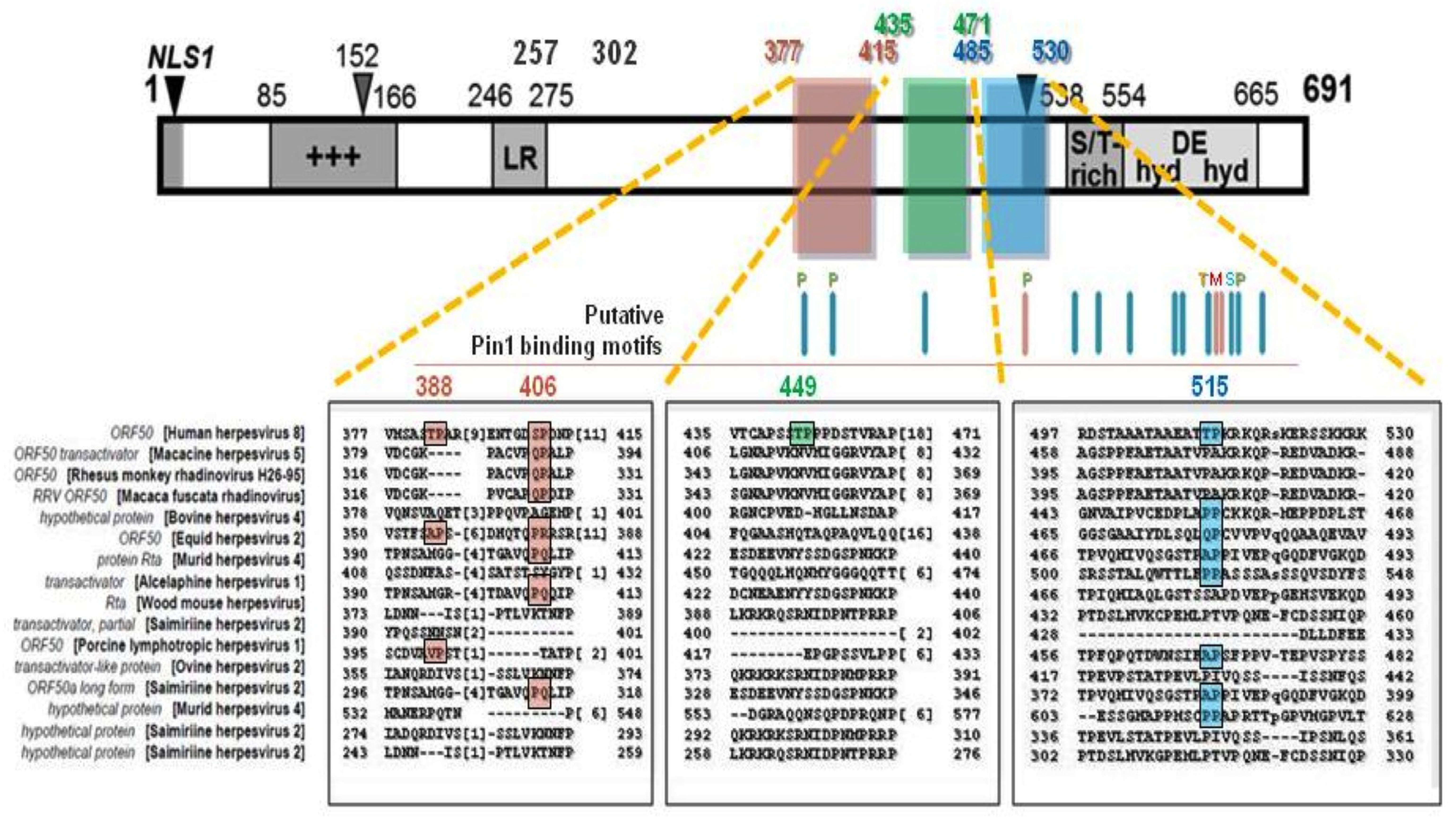
3. Function, Regulation and Dysregulation of Pin1 Isomerase and its Novel Role in KSHV Lytic Reactivation
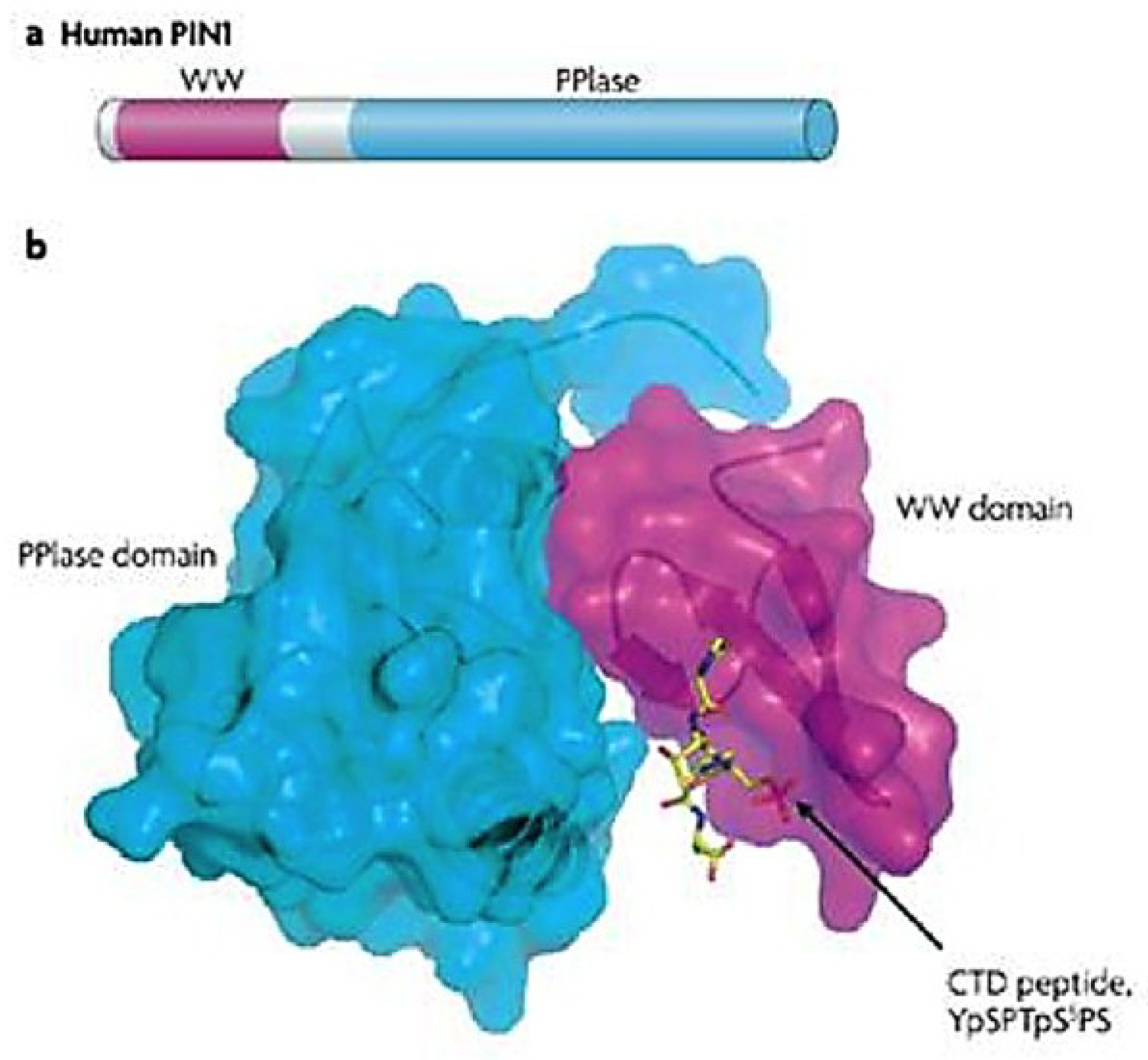
3.1. Human PPIase and Cell Cycle Regulator Pin1


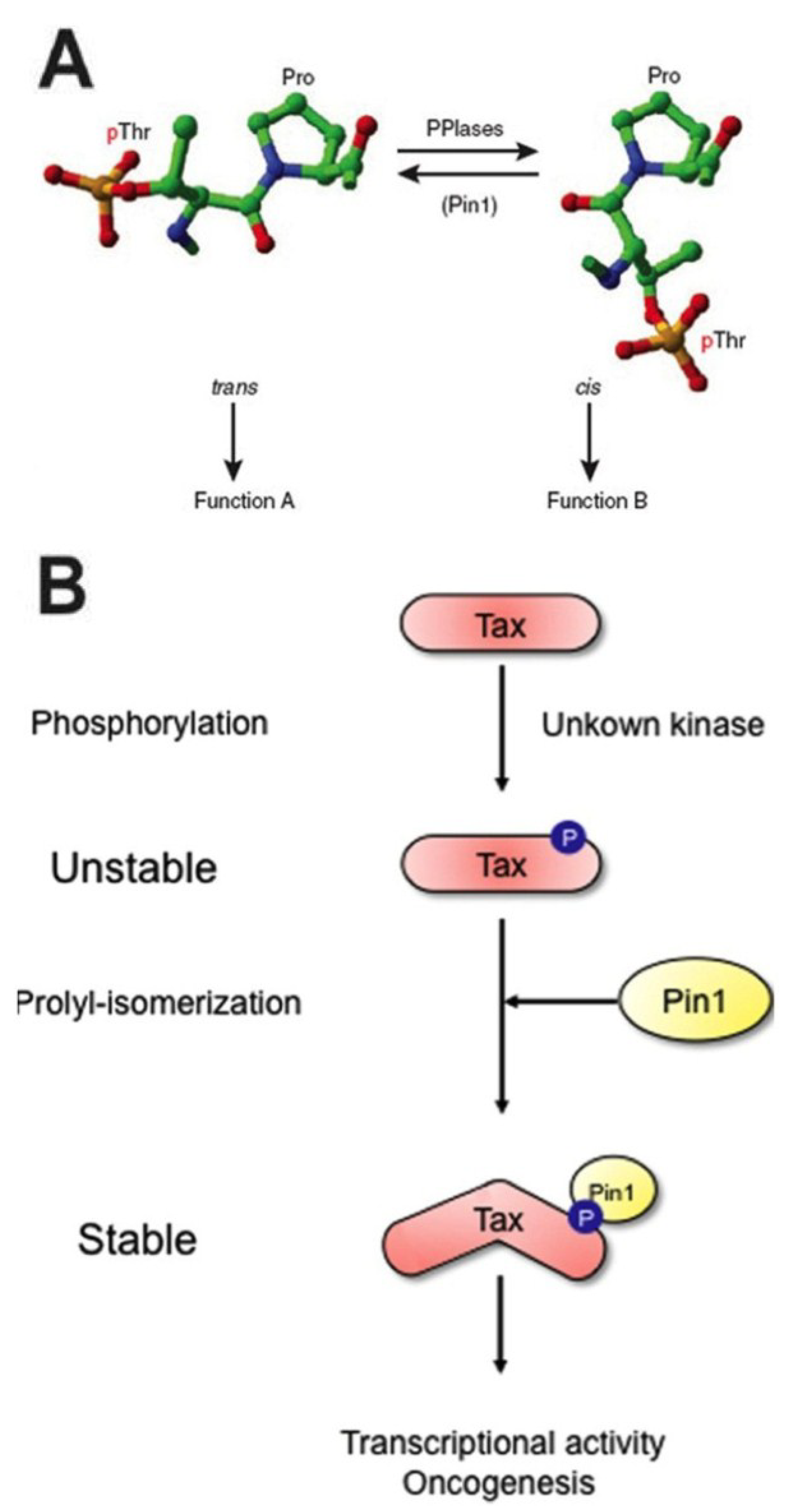
3.2. Dysfunction of Pin1 Is Often Associated with Tumorigenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Substrate Type | Pin1 Interaction | Proposed Pin1 Function |
|---|---|---|---|
| Akt p70S6K | PI3K pathway kinase | Stabilizes/activates |
|
| Cyclin D pRb | G1/S activator G1/S inhibitor | Stabilizes/relocalizes Deactivates |
|
| Pim1 | Oncogenic kinase | Destabilizes |
|
| Raf1 RSK2 | MAPK pathway kinases | + Dephosphor/stabilizes + Phosphor/stabilizes |
|
| SMAD | Transactivator | Reduces protein levels |
|
| Cdc25 Incenp NIMA Survivin TopoIIα Wee1 | Mitotic regulators | Promotes dephosphor Unknown interaction Decreases activity Decreases protein levels Promotes phosphor Deactivates |
|
| Centrosome | Organelle | Enhances activity |
|
| Histone H1 | Chromatin binding protein | + Dephosphor/enhances binding |
|
| Actin Tau | Cytoskeletal proteins | Unknown interaction Promotes dephosphor |
|
| KRMP1 | Kinesin-like motor | Unknown interaction |
|
| c-Myc | TF | Enhances activity/destabilizes |
|
| HDAC3 | Deacetylase | Destabilizes |
|
| SMRT | Transcriptional repressor | Destabilizes |
|
| β-catenin | TF | Stabilizes/activates |
|
| Bcl2 | Antiapoptotic regulator | Destabilizes/deactivates |
|
| c-Jun/c-Fos | TFs | Stabilizes/activates |
|
| p53 | DNA damage response TF | Stabilizes/activates |
|
| p65 (NF-κB) | TF | Relocalizes/stabilizes |
|
| Notch1/NICD | Growth factor receptor | Stimulates cleavage |
|
| Hif-1 BiP/Grp78 | Hypoxia regulator ER stress regulator | Upregulates expression |
|
| APP | Membrane protein | + Dephosphor/destabilizes |
|
| ADAR2 | Adenosine deaminase | Stabilizes |
|
| Nanog Oct4 | Self-renewal TFs | Stabilizes/enhances activity |
|
| RNAP II CTD hSpt5 | Transcriptional regulators | Controls activity/relocalizes |
|
| TRF1 | Shelterin member | Destabilizes |
|
| APOBEC3G Capsid protein Integrase | Cytidine deaminase HIV-1 virion protein HIV-1 enzyme | Inhibits activity Stabilizes Stabilizes |
|
| BALF5 | EBV polymerase catalytic subunit | Enhances activity |
|
| Hbx | HBV transactivator | Stabilizes |
|
| IRF3 | IFN response regulator | Destabilizes homodimers |
|
| Tax | HTLV-1 transactivator | Stabilizes/activates |
|
3.3. Pin1 Has a Novel Role in KSHV Lytic Reactivation

4. Significance of Convergence of Pin1 Function with Regulation of KSHV Lytic Reactivation
4.1. Ectopic Pin1 Is Sufficient to Induce Rta Expression: Putative Mechanisms
4.2. Pin1 Directly Binds to Rta and Enhances Rta Transactivation

4.3. Pin1 Enhances KSHV Lytic DNA Replication
4.4. Pin1 Represses KSHV Late Gene Expression and Virion Production
4.5. Molecular Timing Model for Pin1’s Effects on KSHV Replication and Pathogenesis

Acknowledgements
Author Contributions
Conflicts of Interest
References and Notes
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: Determinants of its pathogenicity? J. Virol. 1997, 71, 4187–4192. [Google Scholar] [PubMed]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Edelman, D.C. Human herpesvirus 8—A novel human pathogen. Virol. J. 2005, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. Kaposi’s sarcoma-associated herpesvirus. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2848–2888. [Google Scholar]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Yuan, Y. Reactivation and lytic replication of KSHV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 434–460. [Google Scholar]
- Ruocco, E.; Ruocco, V.; Tornesello, M.L.; Gambardella, A.; Wolf, R.; Buonaguro, F.M. Kaposi's sarcoma: Etiology and pathogenesis, inducing factors, causal associations, and treatments: facts and controversies. Clin. Dermatol. 2013, 31, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Wang, X.; Yuan, Y. Tegument Proteins of Kaposi’s Sarcoma-Associated Herpesvirus and Related Gamma-Herpesviruses. Front. Microbiol. 2012, 3, 98. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Kuhne, K.; Ye, F.; Chen, J.; Zhou, F.; Lei, X.; Gao, S.J. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat. Res. 2007, 133, 69–127. [Google Scholar] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 2: Pathogenesis, Castleman’s disease, and pleural effusion lymphoma. Lancet Infect. Dis. 2002, 2, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; Rahemtullah, A.; Hochberg, E. Primary effusion lymphoma. Oncologist 2007, 12, 569–576. [Google Scholar] [CrossRef]
- Noguchi, K.; Fukazawa, H.; Murakami, Y.; Takahashi, N.; Yamagoe, S.; Uehara, Y. Gamma-herpesviruses and cellular signaling in AIDS-associated malignancies. Cancer Sci. 2007, 98, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.A.; Harth-Hertle, M.L.; Malterer, G.; Haas, J.; Ellwart, J.; Schulz, T.F.; Kempkes, B. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog. 2013, 9, e1003336. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.-F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Sternbach, G.; Varon, J. Moritz Kaposi: Idiopathic pigmented sarcoma of the skin. J. Emerg. Med. 1995, 13, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Rezza, G.; Lennette, E.T.; Giuliani, M.; Pezzotti, P.; Caprilli, F.; Monini, P.; Butto, S.; Lodi, G.; Di Carlo, A.; Levy, J.A.; Ensoli, B. Prevalence and determinants of anti-lytic and anti-latent antibodies to human herpesvirus-8 among Italian individuals at risk of sexually and parenterally transmitted infections. Int. J. Cancer 1998, 77, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Pantry, S.N.; Medveczky, P.G. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus replication. Semin. Cancer Biol. 2009, 19, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Zhu, F.X.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog. 2009, 5, e1000332. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.; Sehgal, I.; D'Auvergne, O.; Kousoulas, K.G. Kaposi’s sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells. J. Virol. 2010, 84, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Molinolo, A.; Bugge, T.H.; Sawai, E.T.; He, Y.; Li, Y.; Ray, P.E.; Gutkind, J.S. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 2003, 3, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Jung, J.U. Immune evasion in Kaposi’s sarcoma-associated herpes virus associated oncogenesis. Semin. Cancer Biol. 2008, 18, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Areste, C.; Blackbourn, D.J. Modulation of the immune system by Kaposi’s sarcoma-associated herpesvirus. Trends Microbiol. 2009, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Damania, B. Oncogenic gamma-herpesviruses: Comparison of viral proteins involved in tumorigenesis. Nat. Rev. Microbiol. 2004, 2, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, A.D.; Cavallin, L.E.; Vincent, L.; Chiozzini, C.; Eroles, P.; Duran, E.M.; Asgari, Z.; Hooper, A.T.; La Perle, K.M.; Hilsher, C.; Gao, S.J.; Dittmer, D.P.; Rafii, S.; Mesri, E.A. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: A cell and animal model of virally induced Kaposi's sarcoma. Cancer Cell 2007, 11, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Ramsdell, A.K.; Martin, D.; Hu, J.; Sawai, E.T.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi’s sarcoma. Cancer Res. 2006, 66, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.C.; Ciufo, D.M.; Alcendor, D.J.; Wan, X.; Nicholas, J.; Browning, P.J.; Rady, P.L.; Tyring, S.K.; Orenstein, J.M.; Rabkin, C.S.; et al. High-level variability in the ORF-K1 membrane protein gene at the left end of the Kaposi’s sarcoma-associated herpesvirus genome defines four major virus subtypes and multiple variants or clades in different human populations. J. Virol. 1999, 73, 4156–4170. [Google Scholar] [PubMed]
- Polson, A.G.; Wang, D.; DeRisi, J.; Ganem, D. Modulation of host gene expression by the constitutively active G protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2002, 62, 4525–4530. [Google Scholar] [PubMed]
- Lagunoff, M.; Lukac, D.M.; Ganem, D. Immunoreceptor tyrosine-based activation motif-dependent signaling by Kaposi’s sarcoma-associated herpesvirus K1 protein: Effects on lytic viral replication. J. Virol. 2001, 75, 5891–5898. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Boshoff, C.; Weiss, R.A.; Chang, Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996, 274, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Xu, Y.; AuCoin, D.P.; Huete, A.R.; Cei, S.A.; Hanson, L.J.; Pari, G.S. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 2005, 79, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, F.; Xie, J.; Kuhne, K.; Gao, S.J. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 2009, 386, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Khadim, F.; Spadavecchia, S.; Palmeri, D.; Lukac, D.M. Direct interactions of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 2007, 81, 8451–8467. [Google Scholar] [CrossRef] [PubMed]
- Damania, B.; Jeong, J.H.; Bowser, B.S.; DeWire, S.M.; Staudt, M.R.; Dittmer, D.P. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 2004, 78, 5491–5499. [Google Scholar] [CrossRef] [PubMed]
- Kieff, E.; Rickinson, A.B. Epstein-Barr virus and its replication. In Fields Virology, 5th ed.; David, K.M., Ed.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2007. [Google Scholar]
- Campbell, M.; Izumiya, Y. Post-Translational Modifications of Kaposi’s Sarcoma-Associated Herpesvirus Regulatory Proteins—SUMO and KSHV. Front. Microbiol. 2012, 3, 31. [Google Scholar] [PubMed]
- Ko, Y.C.; Tsai, W.H.; Wang, P.W.; Wu, I.L.; Lin, S.Y.; Chen, Y.L.; Chen, J.Y.; Lin, S.F. Suppressive regulation of KSHV RTA with O-GlcNAcylation. J. Biomed. Sci. 2012, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.H.; Wang, P.W.; Lin, S.Y.; Wu, I.L.; Ko, Y.C.; Chen, Y.L.; Li, M.; Lin, S.F. Ser-634 and Ser-636 of Kaposi’s Sarcoma-Associated Herpesvirus RTA are Involved in Transactivation and are Potential Cdk9 Phosphorylation Sites. Front. Microbiol. 2012, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef] [PubMed]
- West, J.T.; Wood, C. The role of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 2003, 22, 5150–5163. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Bu, W.; Palmeri, D.; Spadavecchia, S.; Lynch, S.J.; Marras, S.A.; Tyagi, S.; Lukac, D.M. Kaposi's Sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 2006, 80, 9697–9709. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, J.; Lynch, S.; Lukac, D.M.; Ganem, D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 2002, 16, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ganem, D. Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 8490–8495. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Spadavecchia, S.; Carroll, K.D.; Lukac, D.M. Promoter- and cell-specific transcriptional transactivation by the Kaposi’s sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 2007, 81, 13299–13314. [Google Scholar] [CrossRef] [PubMed]
- Papugani, A.; Coleman, T.; Jones, C.; Zhang, L. The interaction between KSHV RTA and cellular RBP-Jkappa and their subsequent DNA binding are not sufficient for activation of RBP-Jkappa. Virus Res. 2008, 131, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Garibyan, L.; Kirshner, J.R.; Palmeri, D.; Ganem, D. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 2001, 75, 6786–6799. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Carroll, K.D.; Palmeri, D.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 2007, 81, 5788–5806. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Carroll, K.D.; Gonzalez-Lopez, O.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jk/CSL. J. Virol. 2011, 85, 11901–11915. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, J.; Grundhoff, A.; Ganem, D. Exploring the DNA binding interactions of the Kaposi’s sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J. Virol. 2006, 80, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Palmeri, D.; Krishnan, R.; Marin, R.; Aris, V.M.; Soteropoulos, P.; Lukac, D.M. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 2008, 82, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Li, X.; Brown, H.J.; Sun, R. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 2002, 76, 5000–5013. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.; Gradoville, L.; Miller, G. Polyadenylated nuclear RNA encoded by Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1996, 93, 11883–11888. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Darricarrere, N.; Darnell, A.; Myoung, J.; Steitz, J.A. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PloS Pathog. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Chan, M.Y.; Zhu, F.X.; Lukac, D.M.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 2004, 78, 8615–8629. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Hwang, S.; Byun, H.; Lim, C.; Kim, J.W.; Choi, E.J.; Choe, J. Kaposi’s sarcoma-associated herpesvirus open reading frame 50 represses p53-induced transcriptional activity and apoptosis. J. Virol. 2001, 75, 6245–6248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wood, C. The transcriptional repressor K-RBP modulates RTA-mediated transactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6294–6306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wen, H.J.; Minhas, V.; Wood, C. The zinc finger DNA-binding domain of K-RBP plays an important role in regulating Kaposi’s sarcoma-associated herpesvirus RTA-mediated gene expression. Virology 2009, 391, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, S.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.; Chen, L.Y.; Izumiya, C.; Luciw, P.; Kung, H.J. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol. 2003, 77, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Robertson, E. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 2007, 312, 101–136. [Google Scholar] [PubMed]
- Ballestas, M.E.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, F.; Ye, F.; Gao, S.J. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 2008, 379, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; DeCotiis, J.; Giron, M.; Palmeri, D.; Lukac, D.M. Histone deacetylase classes I and II regulate Kaposi’s sarcoma-associated herpesvirus reactivation. J. Virol. 2014, 88, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Jakymiw, A.; Findlay, V.; Parsons, C. KSHV-Encoded MicroRNAs: Lessons for Viral Cancer Pathogenesis and Emerging Concepts. Int. J. Cell Biol. 2012, 2012, 603961. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Ziegelbauer, J. MicroRNAs of Kaposi’s sarcoma-associated herpes virus. Semin. Cancer Biol. 2008, 18, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Xie, J.P.; Kang, T.; Greene, W.; Kuhne, K.; Lei, X.F.; Li, Q.H.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: A novel mechanism of virus control of latency. J. Virol. 2008, 82, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; Hsia, D.; Ellison, T.J.; Luciw, P.A.; Kung, H.J. NF-kappaB serves as a cellular sensor of Kaposi’s sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jkappa coactivator. J. Virol. 2009, 83, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Chen, H.; Shamay, M.; Zheng, Q.; Hayward, G.S. Early activation of the Kaposi’s sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 2004, 78, 4248–4267. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liu, Y.; Liang, D.; Wang, Z.; Robertson, E.S.; Lan, K. Cellular corepressor TLE2 inhibits replication-and-transcription- activator-mediated transactivation and lytic reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 2047–2062. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Miller, G. Autoregulation of DNA binding and protein stability of Kaposi’s sarcoma-associated herpesvirus ORF50 protein. J. Virol. 2004, 78, 10657–10673. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Shedd, D.; Miller, G. A mobile functional region of Kaposi’s sarcoma-associated herpesvirus ORF50 protein independently regulates DNA binding and protein abundance. J. Virol. 2008, 82, 9700–9716. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; Mellinghoff, I.K.; Jamieson, C.; Sun, R. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Weidner-Glunde, M.; Varjosalo, M.; Rainio, E.M.; Lehtonen, A.; Schulz, T.F.; Koskinen, P.J.; Taipale, J.; Ojala, P.M. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Sandford, G.; Choi, Y.B.; Nicholas, J. Role of ORF74-encoded viral G protein-coupled receptor in human herpesvirus 8 lytic replication. J. Virol. 2009, 83, 13009–13014. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Sharma-Walia, N.; Kerur, N.; Paul, A.G.; Sadagopan, S.; Cannon, M.; Chandran, B. Kaposi sarcoma-associated herpes virus (KSHV) G protein-coupled receptor (vGPCR) activates the ORF50 lytic switch promoter: A potential positive feedback loop for sustained ORF50 gene expression. Virology 2009, 392, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Davis, D.A.; Wang, V.; Widmer, I.; Yarchoan, R. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J. Virol. 2003, 77, 6761–6768. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Tsao, E.H.; Webb, B.L.; Ye, H.; Dalton-Griffin, L.; Tsantoulas, C.; Gale, C.V.; Du, M.Q.; Whitehouse, A.; Kellam, P. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 2007, 81, 13578–13586. [Google Scholar] [CrossRef] [PubMed]
- Dalton-Griffin, L.; Wilson, S.J.; Kellam, P. X-box binding protein 1 contributes to induction of the Kaposi’s sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J. Virol. 2009, 83, 7202–7209. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Lin, J.K.; Lee, M.T. Inhibition of 12-O-tetradecanoylphorbol-13-acetate induction of c-fos mRNA by the protein kinase A inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide. Biochem. Pharmacol. 1999, 58, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Hwang, S.; Lim, C.; Won, Y.S.; Lee, C.H.; Choe, J. Kaposi’s Sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 2002, 277, 6438–6442. [Google Scholar] [CrossRef] [PubMed]
- Spadavecchia, S.; Palmeri, D.; Lukac, D.M. KSHV Mta protein stimulates transcriptional elongation. 2009; to be submitted for publication. [Google Scholar]
- Conrad, N.K. Posttranscriptional gene regulation in Kaposi’s sarcoma-associated herpesvirus. Adv. Appl. Microbiol. 2009, 68, 241–261. [Google Scholar] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: Implications for modulating arteriogenesis and angiogenesis. Mol. Cell Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.L.; Reed, L.L.; Broude, E.; Golde, T.E.; Miele, L.; Foreman, K.E. Notch inhibition in Kaposi’s sarcoma tumor cells leads to mitotic catastrophe through nuclear factor-kappaB signaling. Mol. Cancer Ther. 2007, 6, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Tulpule, A.; Zhou, Y.; Scehnet, J.S.; Zhang, S.; Lee, J.S.; Chaudhary, P.M.; Jung, J.; Gill, P.S. KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood 2010, 115, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PloS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.L.; Reed, L.L.; Golde, T.E.; Miele, L.; Nickoloff, B.J.; Foreman, K.E. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 2005, 24, 6333–6344. [Google Scholar] [PubMed]
- Lan, K.; Choudhuri, T.; Murakami, M.; Kuppers, D.A.; Robertson, E.S. Intracellular activated Notch1 is critical for proliferation of Kaposi’s sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 2006, 80, 6411–6419. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Choudhuri, T.; Kuppers, D.A.; Robertson, E.S. Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 2006, 351, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez, O.; Palmeri, D.; Lukac, D.M. Comprehensive analysis of DNA sequences required for KSHV Rta to stimulate DNA binding of the Notch effector RBP-Jk. 2015; to be submitted for publication. [Google Scholar]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.J. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Spadavecchia, S.; Gonzalez-Lopez, O.; Carroll, K.D.; Palmeri, D.; Lukac, D.M. Convergence of Kaposi’s sarcoma-associated herpesvirus reactivation with Epstein-Barr virus latency and cellular growth mediated by the notch signaling pathway in coinfected cells. J. Virol. 2010, 84, 10488–10500. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res. 2012, 40, 4273–4287. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. The structure and function of proline-rich regions in proteins. Biochem. J. 1994, 297, 249–260. [Google Scholar] [PubMed]
- Chang, M.; Brown, H.J.; Collado-Hidalgo, A.; Arevalo, J.M.; Galic, Z.; Symensma, T.L.; Tanaka, L.; Deng, H.; Zack, J.A.; Sun, R.; Cole, S.W. beta-Adrenoreceptors reactivate Kaposi’s sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA. J. Virol. 2005, 79, 13538–13547. [Google Scholar] [CrossRef] [PubMed]
- Gothel, S.F.; Marahiel, M.A. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol. Life Sci. 1999, 55, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.X. Protein folding. Prolyl isomerases join the fold. Curr. Biol. 1995, 5, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Dolinski, K.; Muir, S.; Cardenas, M.; Heitman, J. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Rahfeld, J.; Fischer, G.; Schmid, F.X. Catalysis of protein folding by parvulin. J. Mol. Biol. 1997, 273, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, M.Y.; Watts, R.E.; Tan, Z.; Cornish, V.W.; Ehrenberg, M.; Forster, A.C. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl. Acad. Sci. USA 2009, 106, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Tsai, F.T. Molecular chaperones in protein quality control. J. Biochem. Mol. Biol. 2005, 38, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem. Sci. 2004, 29, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Frausto, S.D.; Lee, E.; Tang, H. Cyclophilins as modulators of viral replication. Viruses 2013, 5, 1684–1701. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Hanes, S.D.; Hunter, T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [CrossRef] [PubMed]
- O'Connell, M.J.; Krien, M.J.; Hunter, T. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 2003, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Haggblom, C.; Vogt, M.; Hunter, T.; Lu, K.P. Characterization and cell cycle regulation of the related human telomeric proteins Pin2 and TRF1 suggest a role in mitosis. Proc. Natl. Acad. Sci. USA 1997, 94, 13618–13623. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Perrem, K.; Harper, J.W.; Lu, K.P.; Zhou, X.Z. The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J. Biol. Chem. 2006, 281, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Uchida, C.; Shin, R.W.; Shimazaki, K.; Uchida, T. Prolyl isomerase, Pin1: New findings of post-translational modifications and physiological substrates in cancer, asthma and Alzheimer’s disease. Cell Mol Life Sci. 2008, 65, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Hanes, S.D. The Ess1 prolyl isomerase: Traffic cop of the RNA polymerase II transcription cycle. Biochim. Biophys. Acta 2014, 1839, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Scherer, G.; Mayr, L.M.; Schindler, T.; Fischer, G.; Schmid, F.X. Prolyl isomerases do not catalyze isomerization of non-prolyl peptide bonds. Biol. Chem. 1998, 379, 361–365. [Google Scholar] [PubMed]
- Lu, K.P.; Liou, Y.C.; Zhou, X.Z. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002, 12, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Ryo, A. Pinning down viral proteins: A new prototype for virus-host cell interaction. Front. Microbiol. 2010, 1, 107. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.; Finn, G.; Suizu, F.; Lu, K.P. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Biol. 2005, 7, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.G. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Ryo, A.; Huang, H.K.; Lu, P.J.; Bronson, R.; Fujimori, F.; Uchida, T.; Hunter, T.; Lu, K.P. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. USA 2002, 99, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Hirose, Y.; Zhou, X.Z.; Lu, K.P.; Manley, J.L. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003, 17, 2765–2776. [Google Scholar] [CrossRef] [PubMed]
- Wildemann, D.; Hernandez Alvarez, B.; Stoller, G.; Zhou, X.Z.; Lu, K.P.; Erdmann, F.; Ferrari, D.; Fischer, G. An essential role for Pin1 in Xenopus laevis embryonic development revealed by specific inhibitors. Biol. Chem. 2007, 388, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Tun-Kyi, A.; Shi, R.; Lim, J.; Soohoo, C.; Finn, G.; Balastik, M.; Pastorino, L.; Wulf, G.; Zhou, X.Z.; Lu, K.P. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat. Cell Biol. 2009, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Raghuram, N.; Strickfaden, H.; McDonald, D.; Williams, K.; Fang, H.; Mizzen, C.; Hayes, J.J.; Th'ng, J.; Hendzel, M.J. Pin1 promotes histone H1 dephosphorylation and stabilizes its binding to chromatin. J. Cell Biol. 2013, 203, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Titus, M.A.; Thapar, R. The prolyl isomerase pin1 regulates mRNA levels of genes with short half-lives by targeting specific RNA binding proteins. PLoS One 2014, 9, e85427. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.; Garg, P.; Liou, Y.C.; Iglehart, D.; Lu, K.P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004, 23, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Cho, Y.G.; Park, Y.G.; Nam, S.W.; Kim, S.Y.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, W.S. Pin1 overexpression in colorectal cancer and its correlation with aberrant beta-catenin expression. World J. Gastroenterol. 2005, 11, 5006–5009. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Uemura, H.; Ishiguro, H.; Saitoh, T.; Yamaguchi, A.; Perrem, K.; Kubota, Y.; Lu, K.P.; Aoki, I. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin. Cancer Res. 2005, 11, 7523–7531. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, J.; Feng, W.; Zhang, S.; Liang, H.; Wang, Y.; Zheng, Q.; Li, Z. PIN1 gene overexpression and beta-catenin gene mutation/expression in hepatocellular carcinoma and their significance. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2007, 27, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.R.; Choi, H.S.; Heo, T.H.; Hwang, S.W.; Kang, K.W. Induction of vascular endothelial growth factor by peptidyl-prolyl isomerase Pin1 in breast cancer cells. Biochem. Biophys. Res. Commun. 2008, 369, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Rustighi, A.; Tiberi, L.; Soldano, A.; Napoli, M.; Nuciforo, P.; Rosato, A.; Kaplan, F.; Capobianco, A.; Pece, S.; Di Fiore, P.P.; Del Sal, G. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 2009, 11, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Kim, G.; Yun, H.J.; Cho, H.G.; Choi, H.S. The prolyl isomerase Pin1 interacts with and downregulates the activity of AMPK leading to induction of tumorigenicity of hepatocarcinoma cells. Mol. Carcinog. 2013, 52, 813–823. [Google Scholar] [PubMed]
- Suizu, F.; Ryo, A.; Wulf, G.; Lim, J.; Lu, K.P. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol. Cell Biol. 2006, 26, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.P.; Nozell, S.E.; Harrison, D.K.; Stonecypher, M.S.; Chen, D.; Benveniste, E.N. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene 2009, 28, 3735–3745. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Choi, H.K.; Shim, J.H.; Kang, K.W.; Dong, Z.; Choi, H.S. The prolyl isomerase Pin1 interacts with a ribosomal protein S6 kinase to enhance insulin-induced AP-1 activity and cellular transformation. Carcinogenesis 2009, 30, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Pulikkan, J.A.; Dengler, V.; Peer Zada, A.A.; Kawasaki, A.; Geletu, M.; Pasalic, Z.; Bohlander, S.K.; Ryo, A.; Tenen, D.G.; Behre, G. Elevated PIN1 expression by C/EBPalpha-p30 blocks C/EBPalpha-induced granulocytic differentiation through c-Jun in AML. Leukemia 2010, 24, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Wang, D.; Wulf, G.; Frolov, A.; Li, R.; Sowadski, J.; Wheeler, T.M.; Lu, K.P.; Bao, L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003, 63, 6244–6251. [Google Scholar] [PubMed]
- Dochi, T.; Nakano, T.; Inoue, M.; Takamune, N.; Shoji, S.; Sano, K.; Misumi, S. Phosphorylation of human immunodeficiency virus type 1 (HIV-1) capsid protein at serine 16, required for peptidyl-prolyl isomerase (Pin1)-dependent uncoating, is mediated by virion-incorporated extracellular signal-regulated kinase 2 (ERK2). J. Gen. Virol. 2014, 95, 1156–1156. [Google Scholar] [CrossRef] [PubMed]
- Misumi, S.; Inoue, M.; Dochi, T.; Kishimoto, N.; Hasegawa, N.; Takamune, N.; Shoji, S. Uncoating of human immunodeficiency virus type 1 requires prolyl isomerase Pin1. J. Biol. Chem. 2010, 285, 25185–25195. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Khan, M.; Yedavalli, V.R.; Yeung, M.L.; Strebel, K.; Jeang, K.T. Human immunodeficiency virus type 1 replication and regulation of APOBEC3G by peptidyl prolyl isomerase Pin1. J. Virol. 2008, 82, 9928–9936. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Tran, H.T.; Park, S.J.; Yim, S.A.; Hwang, S.B. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J. Virol. 2011, 85, 8777–8788. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Lu, K.P. Hepatitis B virus x protein and pin1 in liver cancer: “les liaisons dangereuses”. Gastroenterology 2007, 132, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.H.; Radonovich, M.; Mahieux, R.; Pise-Masison, C.; Muralidhar, S.; Brady, J.N. P53 facilitates degradation of human T-cell leukaemia virus type I Tax-binding protein through a proteasome-dependent pathway. J. Gen. Virol. 2003, 84, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Peloponese, J.M., Jr.; Yasunaga, J.; Kinjo, T.; Watashi, K.; Jeang, K.T. Peptidylproline cis-trans-isomerase Pin1 interacts with human T-cell leukemia virus type 1 tax and modulates its activation of NF-kappaB. J. Virol. 2009, 83, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Johnson, D.G.; Kane, B.P.; Sowder, R.C., 2nd; Kim, Y.D.; Fisher, R.J.; Zhou, X.Z.; Lu, K.P.; Henderson, L.E. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 2000, 266, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Kamimoto, T.; Zama, T.; Aoki, R.; Muro, Y.; Hagiwara, M. Identification of a novel kinesin-related protein, KRMP1, as a target for mitotic peptidyl-prolyl isomerase Pin1. J. Biol. Chem. 2001, 276, 37520–37528. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Murata, T.; Ryo, A.; Kawashima, D.; Sugimoto, A.; Kanda, T.; Kimura, H.; Tsurumi, T. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J. Virol. 2013, 87, 2120–2127. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Gavina, A.; Palmeri, D.; Lukac, D.M. The cellular peptidyl-prolyl cis/trans isomerase Pin1 regulates reactivation of Kaposi’s sarcoma-associated herpesvirus from latency. J. Virol. 2014, 88, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Carlsson, J.; Ikoma, M.; Gachelet, E.; Gray, M.; Geballe, A.P.; Corey, L.; Casper, C.; Lagunoff, M.; Vieira, J. The HIV protease inhibitor nelfinavir inhibits Kaposi’s sarcoma-associated herpesvirus replication in vitro. Antimicrob. Agents Chemother. 2011, 55, 2696–2703. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.F.; Wong, L.Y.; Lee, H.R.; Chung, B.; Jung, J.U. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. J. Virol. 2012, 86, 9696–9707. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Bi, M.; Ye, J.; Feldman, D.; Koong, A.C. Hypoxia and the unfolded protein response. Methods Enzymol. 2007, 435, 275–293. [Google Scholar] [PubMed]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Long, E.; Ilie, M.; Hofman, V.; Havet, K.; Selva, E.; Butori, C.; Lacour, J.P.; Nelson, A.M.; Cathomas, G.; Hofman, P. LANA-1, Bcl-2, Mcl-1 and HIF-1alpha protein expression in HIV-associated Kaposi sarcoma. Virchows Arch. 2009, 455, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.R.; Choi, H.S.; Yang, J.W.; Park, B.C.; Kim, J.A.; Kang, K.W. Enhancement of vascular endothelial growth factor-mediated angiogenesis in tamoxifen-resistant breast cancer cells: Role of Pin1 overexpression. Mol. Cancer Ther. 2009, 8, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Park, S.Y.; Kim, D.J.; Lee, S.H.; Woo, K.M.; Lee, K.A.; Lee, Y.J.; Cho, Y.Y.; Shim, J.H. TPA-induced cell transformation provokes a complex formation between Pin1 and 90 kDa ribosomal protein S6 kinase 2. Mol. Cell Biochem. 2012, 367, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Manley, J.L. Pinning down transcription: Regulation of RNA polymerase II activity during the cell cycle. Cell Cycle 2004, 3, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Manley, J.L. Pin1 modulates RNA polymerase II activity during the transcription cycle. Genes Dev. 2007, 21, 2950–2962. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.M.; Liou, Y.C.; Ryo, A.; Lee, S.W.; Lu, K.P. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 2002, 277, 47976–47979. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wulf, G.; Gu, L.; Tang, X.; Lu, K.P.; Xiao, Z.X. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wei, Y.; Zhou, X.; Yang, J.Y.; Dai, C.; Chen, Y.J.; Agarwal, N.K.; Sarbassov, D.; Shi, D.; Yu, D.; Hung, M.C. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 2009, 28, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Zhou, X.Z.; Liou, Y.C.; Noel, J.P.; Lu, K.P. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J. Biol. Chem. 2002, 277, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, S.B.; Albert, A.L.; Handa, H.; Vincent, M.; Bensaude, O. The peptidyl-prolyl isomerase Pin1 interacts with hSpt5 phosphorylated by Cdk9. J. Mol. Biol. 2001, 312, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Verma, S.C.; Murakami, M.; Bajaj, B.; Kaul, R.; Robertson, E.S. Kaposi’s sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc. Natl. Acad. Sci. USA 2007, 104, 16287–16292. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.R.; Lukac, D.M.; Chang, J.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 2000, 74, 3586–3597. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guito, J.; Lukac, D.M. KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control. Viruses 2015, 7, 72-109. https://doi.org/10.3390/v7010072
Guito J, Lukac DM. KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control. Viruses. 2015; 7(1):72-109. https://doi.org/10.3390/v7010072
Chicago/Turabian StyleGuito, Jonathan, and David M. Lukac. 2015. "KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control" Viruses 7, no. 1: 72-109. https://doi.org/10.3390/v7010072
APA StyleGuito, J., & Lukac, D. M. (2015). KSHV Reactivation and Novel Implications of Protein Isomerization on Lytic Switch Control. Viruses, 7(1), 72-109. https://doi.org/10.3390/v7010072