
Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants



Abstract
:
1. Introduction
2. Results
2.1. Site Selection

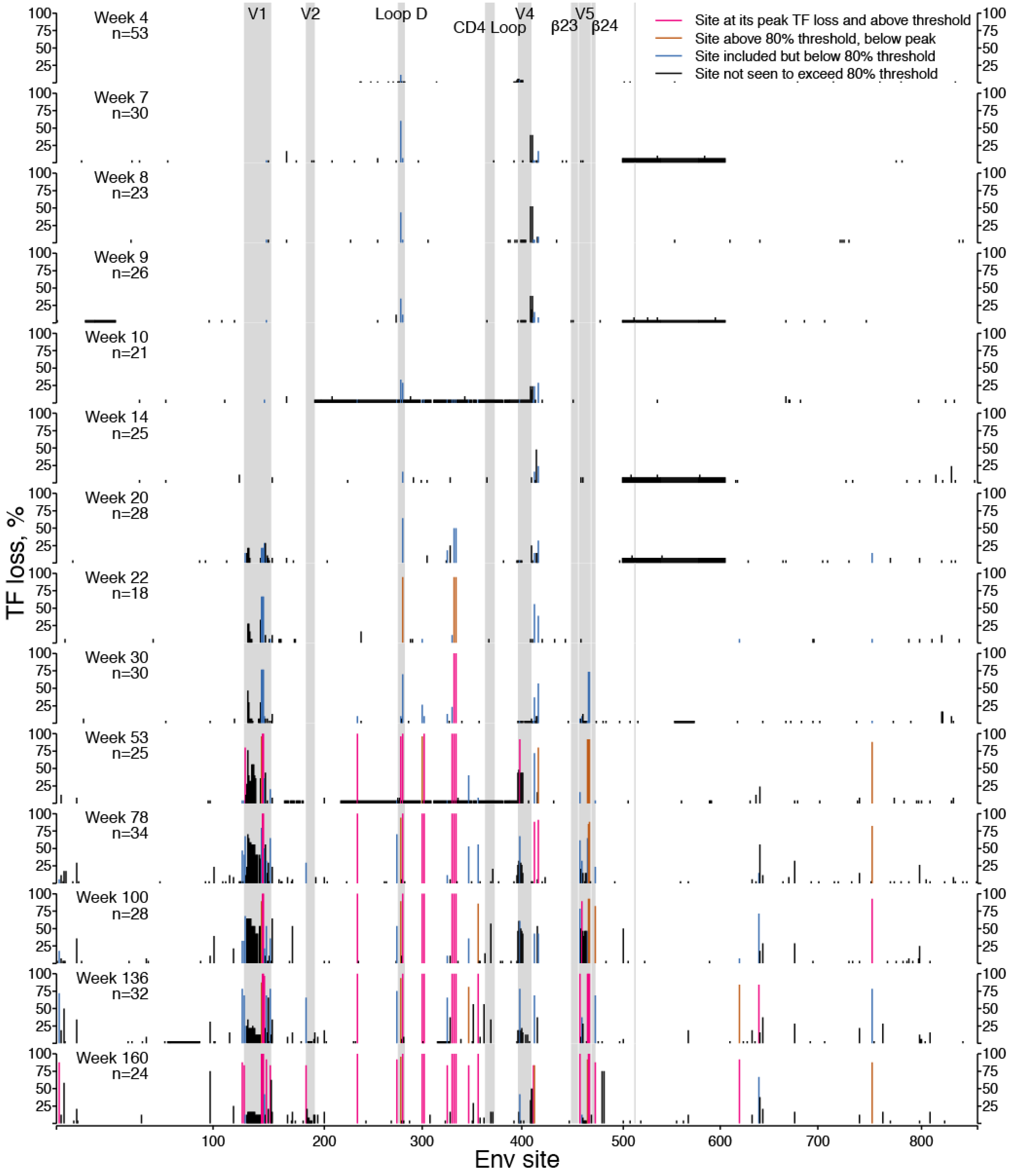
2.1.1. TF Loss Varied across Sites
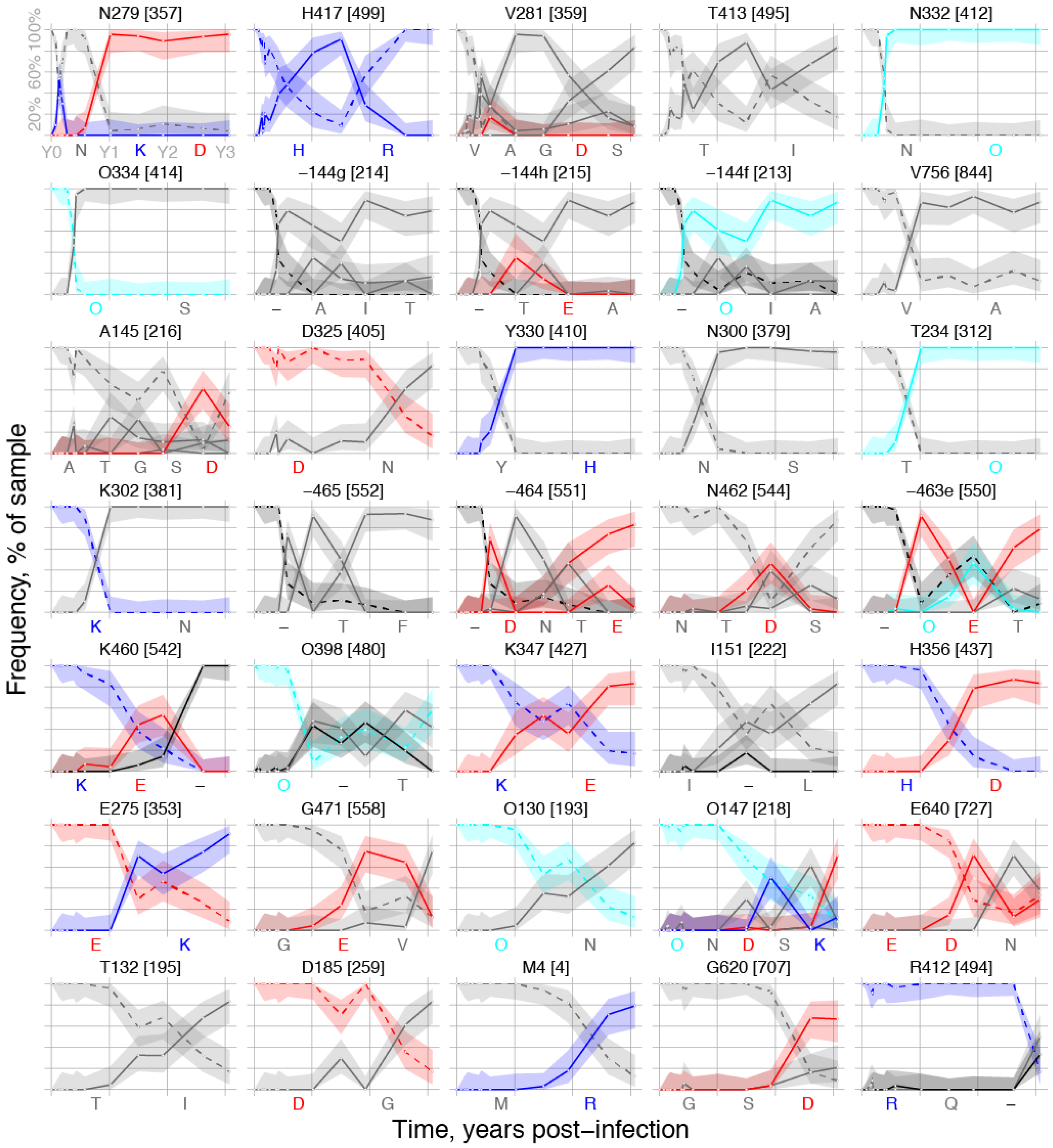
2.1.2. Peak TF Loss Identified Selected Sites


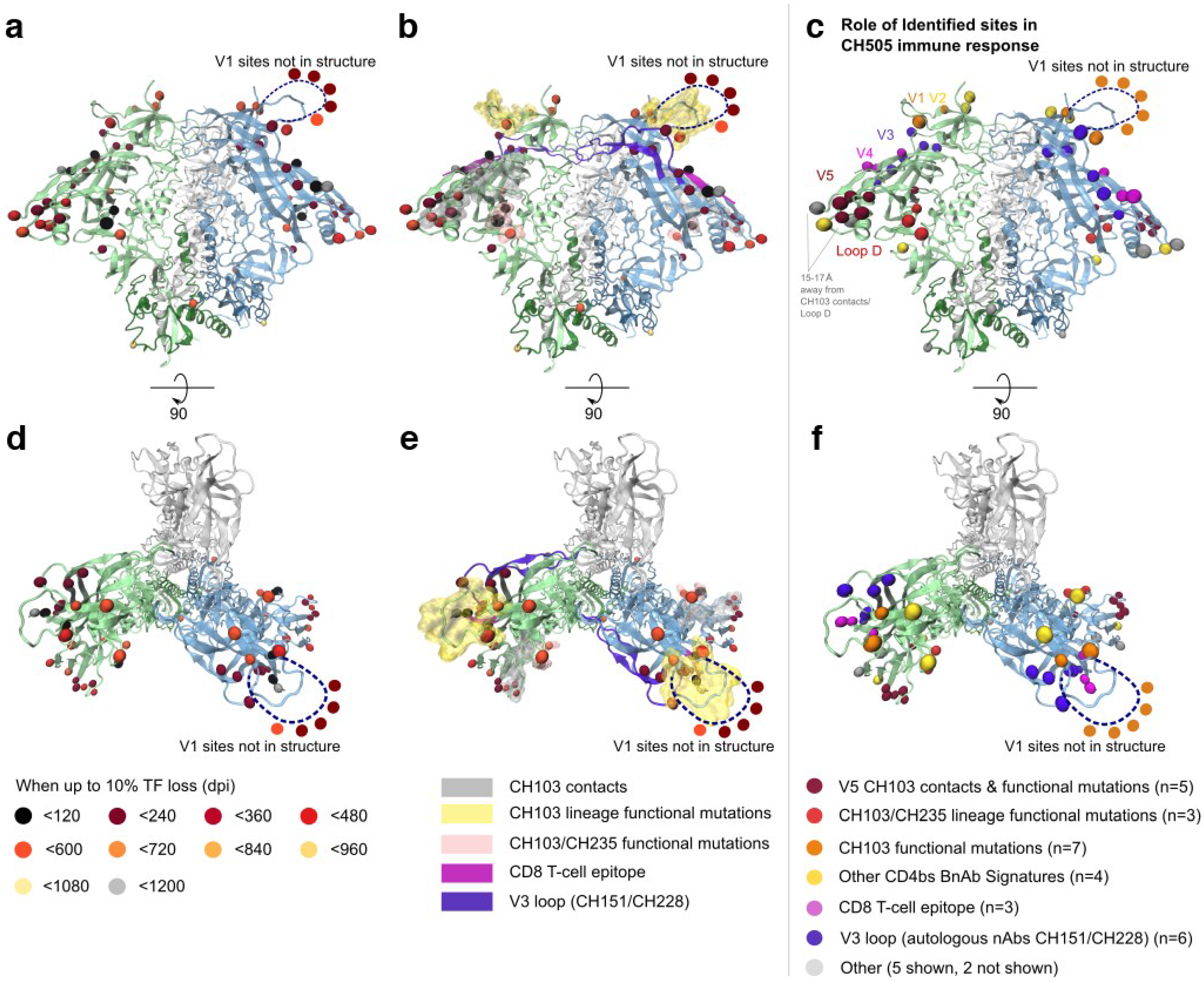
2.1.3. Selected Sites were Consistent with Antibody-Driven Selection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HXB2 Site | Peak Loss | When Up | Rank | MEME p-Value | Immune Pressure | Notes | |
|---|---|---|---|---|---|---|---|
| 4 | 87.5 | d701 | 33 | NA 1 | NA | Signal peptide | |
| 2 | 130 | 87.5 | d547 | 28 | 0.086 | CD4bs | PNG site at base of V1, near VRC01 contact [46] |
| 132 | 83.3 | d547 | 31 | * 3 | CD4bs | V1 indels cause CH103 resistance [16] | |
| 144f | 100 | d141 | 9 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 144g | 100 | d141 | 7 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 144h | 100 | d141 | 8 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 145 | 96.8 | d141 | 11 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 147 | 91.7 | d547 | 29 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 151 | 83.3 | d371 | 24 | * | CD4bs | V1 indels cause CH103 resistance [16] | |
| 185 | 83.3 | d547 | 32 | 0.013 | CD4bs | Signature site for CD4bs bnAb b12 [47] | |
| 234 | 100 | d211 | 15 | NA | CD4bs | Signature site for CD4bs VRC01 & NIH45-56 [47] | |
| 275 | 91.7 | d547 | 26 | 0.044 | CD4bs | Loop D, CH103 contact, CH235 resistant [15,16] | |
| 279 | 95.8 | d28 | 1 | 0.090 | CD4bs | Loop D, CH235 resistant, CH103 sensitive [15,16] | |
| 281 | 100 | d64 | 3 | 0.00007 4 | CD4bs | Loop D, CH235 resistant, CH103 sensitive [15,16] | |
| 300 | 100 | d211 | 14 | NA | V3 loop | V3 autologous nAb in CH505 [41] | |
| 302 | 100 | d211 | 16 | NA | V3 loop | V3 autologous nAb in CH505 [41] | |
| 325 | 83.3 | d141 | 12 | NA | V3 loop | V3 autologous nAb in CH505 [41] | |
| 330 | 100 | d157 | 13 | NA | V3 loop | V3 autologous nAb in CH505 [41] | |
| 332 | 100 | d141 | 5 | V3 loop | V3 autologous nAb in CH505 [41] | ||
| 334 | 100 | d141 | 6 | 0.029 | V3 loop | V3 autologous nAb in CH505 [41] | |
| 347 | 83.3 | d371 | 23 | 0.0074 | CD4bs | 15–17 Angstroms from CH103 contacts | |
| 356 | 100 | d547 | 25 | 0.018 | CD4bs | Adjacent to CD4bs bnAb 12A12 signature [47] | |
| 398 | 91.3 | d371 | 22 | 0.0088 | CD4bs | 15-17 Angstroms from CH103 contacts | |
| 412 | 83.3 | d1121 | 35 | NA | CTL | CTL epitope V4 loop [16] | |
| 413 | 88.2 | d64 | 4 | 0.00004 | CTL | CTL epitope V4 loop [16] | |
| 417 | 91.2 | d51 | 2 | 0.00073 | CTL/CD4bs | CTL epitope V4 loop; CD4bs b12 contact [16] | |
| 460 | 100 | d371 | 21 | * | CD4bs | V5, CH103 contact region, resistance [15,16] | |
| 462 | 89.3 | d211 | 19 | * | CD4bs | V5, CH103 contact region, resistance [15,16] | |
| 463e | 100 | d371 | 20 | * | CD4bs | V5, CH103 contact region, resistance [15,16] | |
| 464 | 100 | d211 | 18 | * | CD4bs | V5, CH103 contact region, resistance [15,16] | |
| 465 | 100 | d211 | 17 | * | CD4bs | V5, CH103 contact region, resistance [15,16] | |
| 471 | 87.5 | d547 | 27 | 0.0057 | CD4bs | CH103 contact [16] | |
| 620 | 91.7 | d953 | 34 | 0.0026 | NA | gp41 | |
| 640 | 83.9 | d547 | 30 | 0.0054 | NA | gp41 | |
| 756 | 92.9 | d141 | 10 | 0.0035 | NA | gp41 cytoplasmic tail |

2.1.4. Comparison of Selected Sites Identified by LASSIE and Phylogenetic Methods
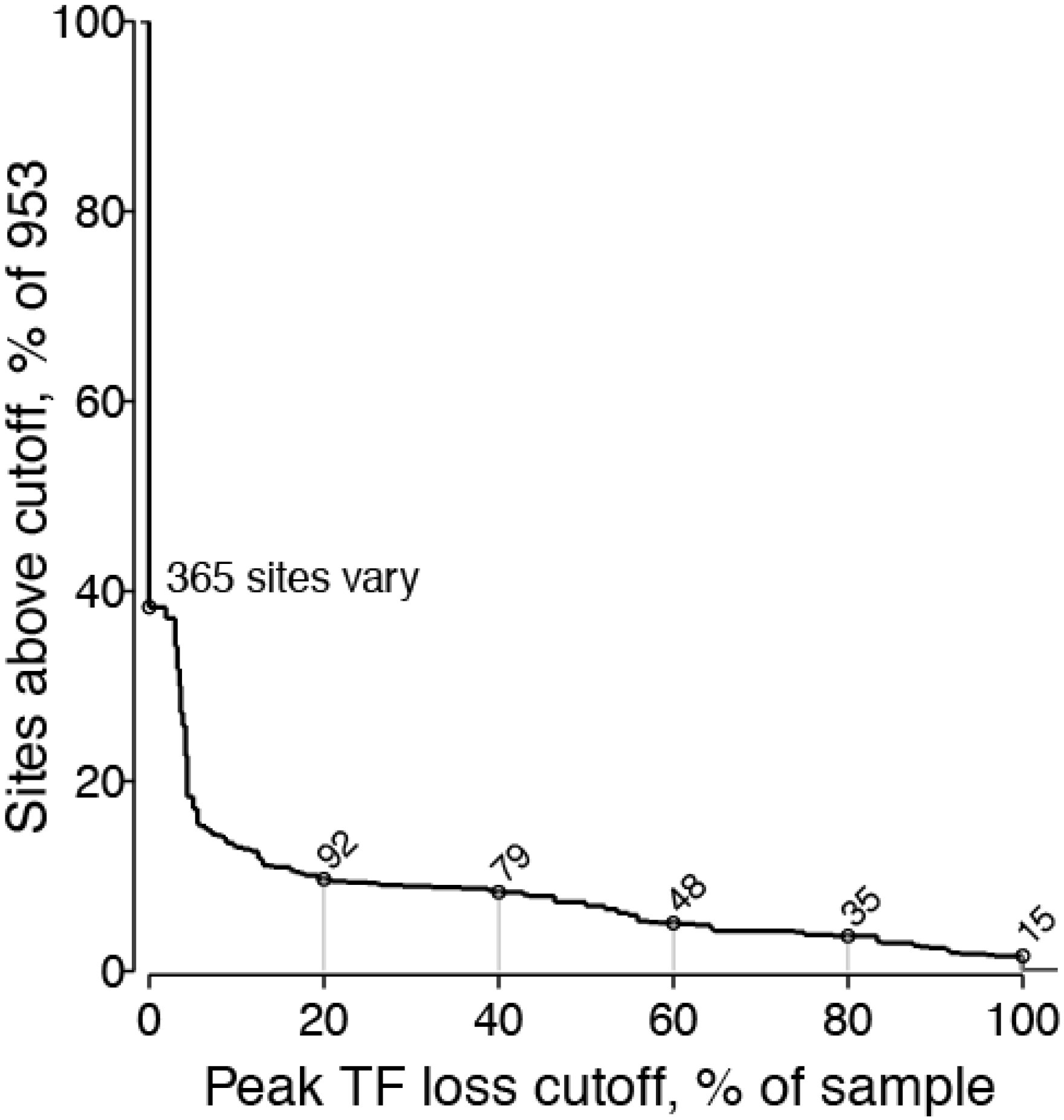
2.1.5. Threshold Considerations
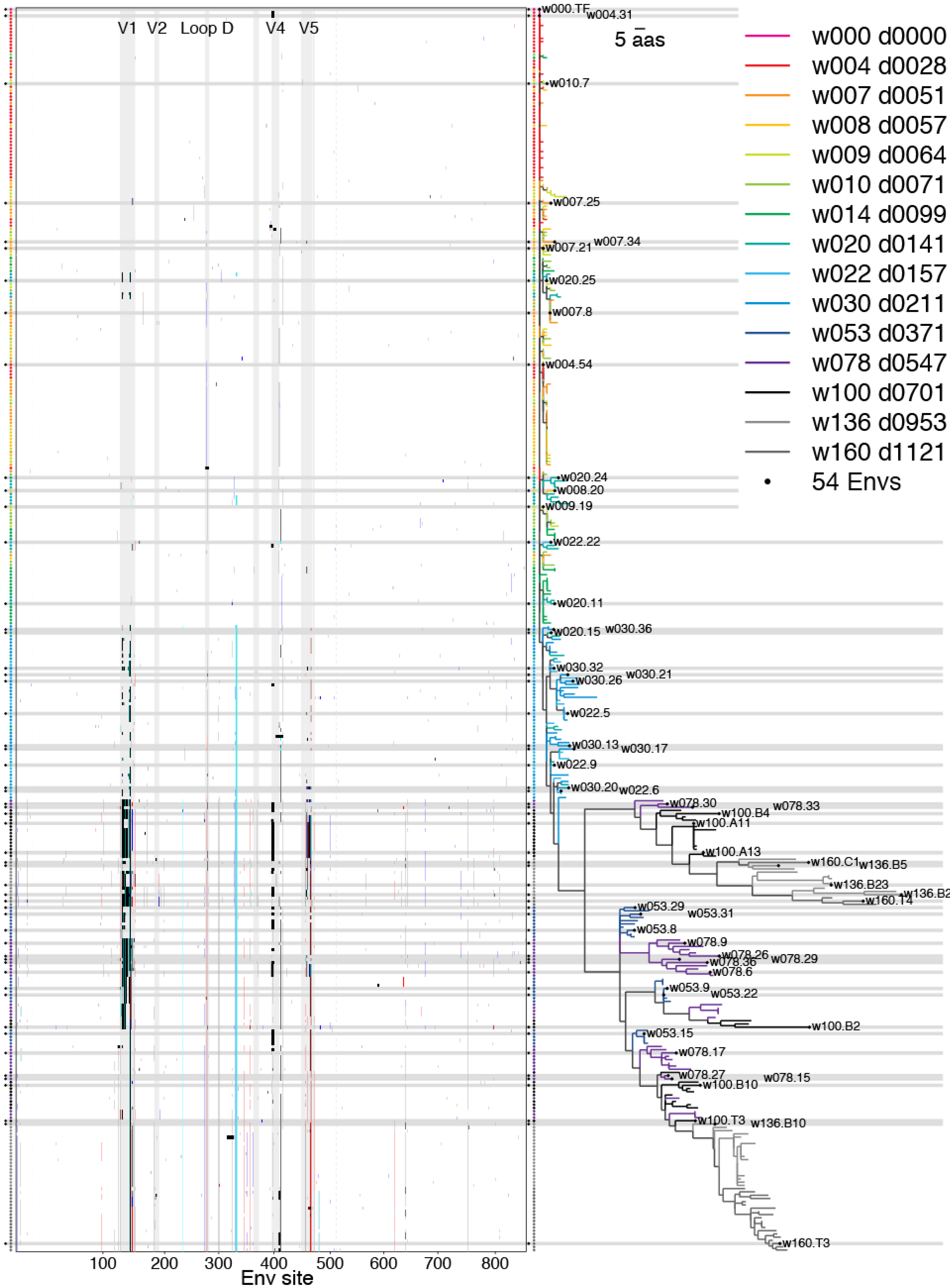
2.1.6. Concatamers of Selected Sites

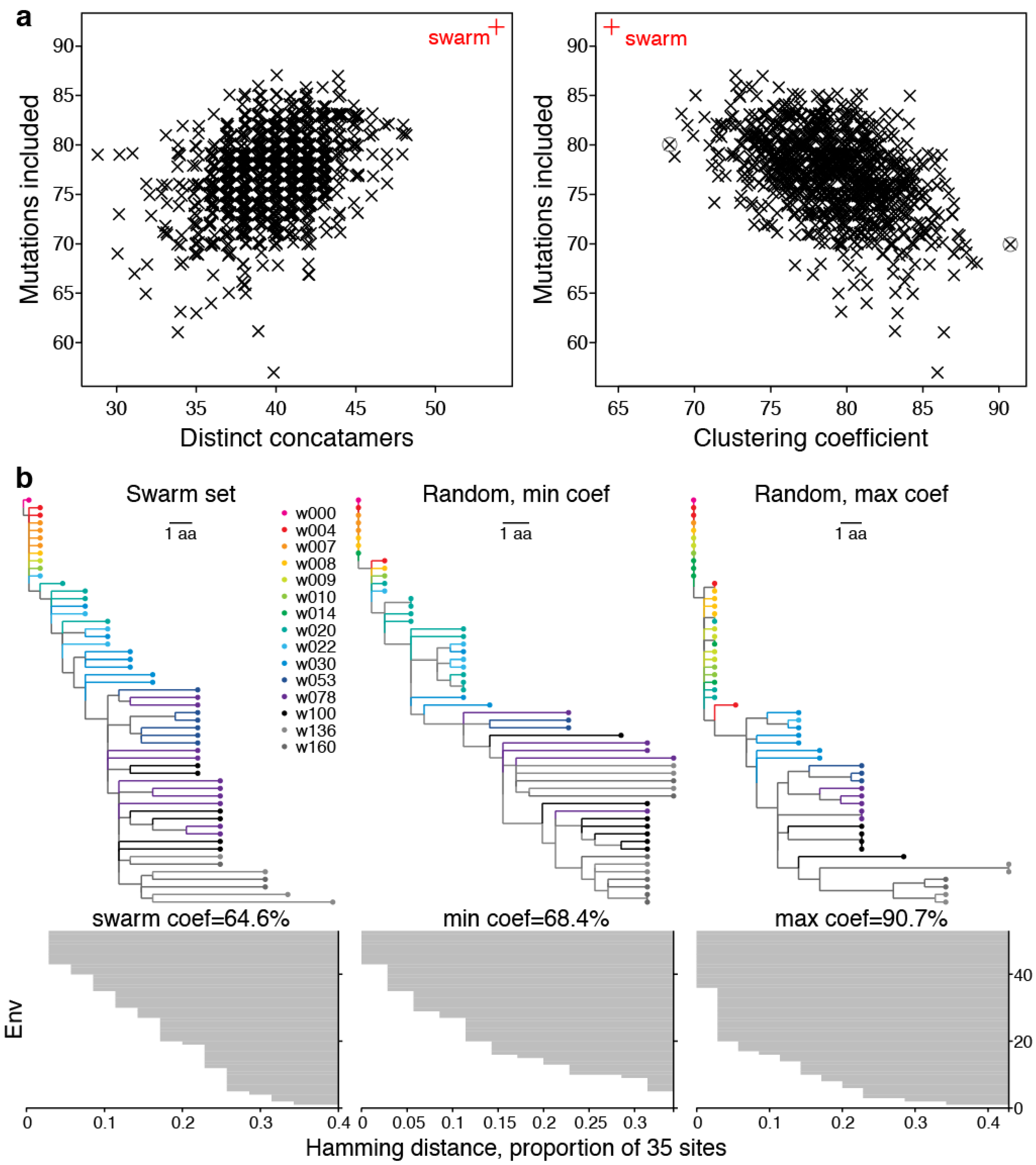
2.2. Swarm Selection
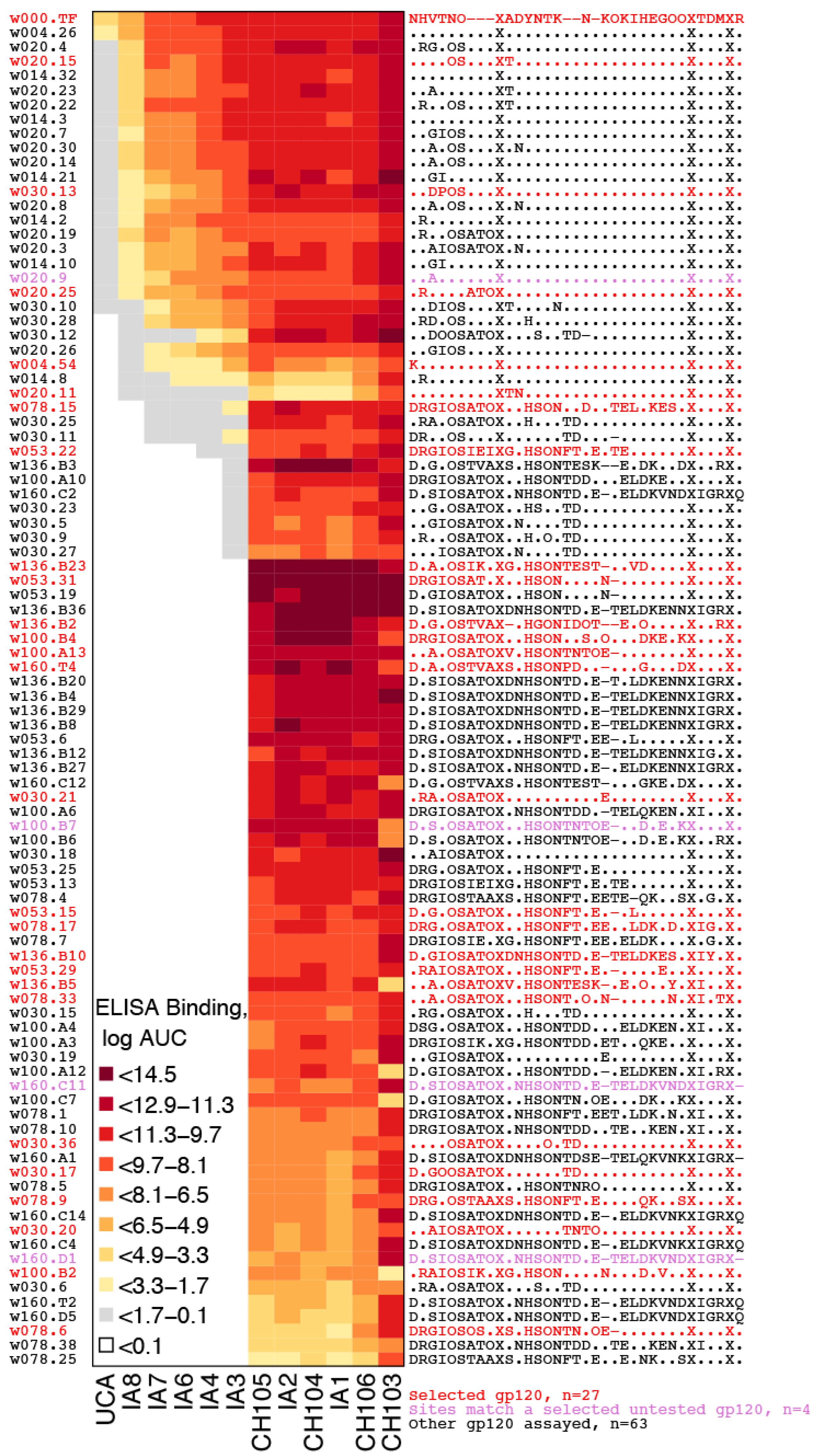
2.2.1. Representative Variants among Selected Sites
2.2.2. LASSIE Compared with Randomly Selected Sequences

2.2.3. Phylogenetic Context

2.2.4. Antigenic Diversity

2.3. Swarm Size Adjustments
2.4. Chronic Infection
3. Discussion
3.1. Site Selection
3.2. Sequence Selection
4. Materials and Methods
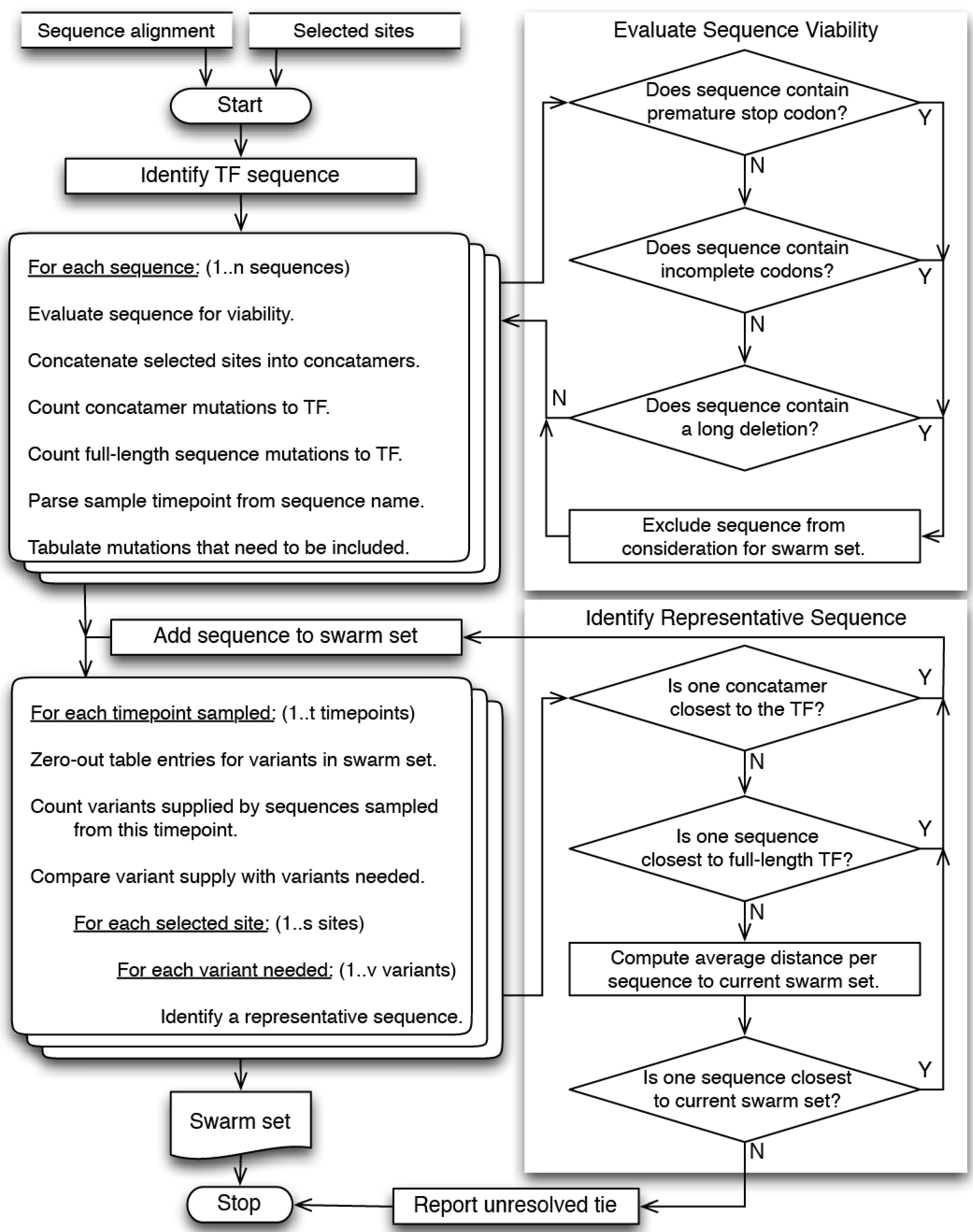
4.1. Overview
4.2. Site Selection
4.3. Sequence Selection

4.4. Availability
4.5. Positively Selected Sites by MEME and FEL Analyses
4.6. Clinical Sample Assays
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plotkin, S.A. Correlates of vaccine-induced immunity. Clin. Infect. Dis. 2008, 47, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.M.; Montefiori, D.M. The role of antibodies in HIV vaccines. Annu. Rev. Immunol. 2010, 28, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.R.; Lewis, M.G.; Stiegler, G.; Harris, D.; VanCott, T.C.; Hayes, D.; Louder, M.K.; Brown, C.R.; Sapan, C.V.; Frankel, S.S.; et al. Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 1999, 73, 4009–4018. [Google Scholar] [PubMed]
- Moldt, B.; Rakasz, E.G.; Schultz, N.; Chan-Hui, P.Y.; Swiderek, K.; Weisgrau, K.L.; Piaskowski, S.M.; Bergman, Z.; Watkins, D.I.; Poignard, P.; et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18921–18925. [Google Scholar] [PubMed]
- Keele, B.; Giorgi, E.; Salazar-Gonzalez, J.; Decker, J.; Pham, K.; Salazar, M.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, E.; Funkhouser, B.; Athreya, G.; Perelson, A.; Korber, B.; Bhattacharya, T. Estimating time since infection in early homogeneous HIV-1 samples using a Poisson model. BMC Bioinformatics 2010, 11, 532. [Google Scholar] [CrossRef] [PubMed]
- Mellors, J.W.; Rinaldo, C.R., Jr.; Gupta, P.; White, R.M.; Todd, J.A.; Kingsley, L.A. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 1996, 272, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Mackelprang, R.D.; Carrington, M.; Thomas, K.K.; Hughes, J.P.; Baeten, J.M.; Wald, A.; Farquhar, C.; Fife, K.; Campbell, M.S.; Kapiga, S.; et al. Host genetic and viral determinants of HIV-1 RNA set point among HIV-1 seroconverters from sub-Saharan Africa. J. Virol. 2015, 89, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, S.M.; Korber, B.T.; Neumann, A.U.; Daniels, M.; Kunstman, K.J.; Whetsell, A.J.; Furtado, M.R.; Cao, Y.; Ho, D.D.; Safrit, J.T. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 1996, 272, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Clapham, P.; Weber, J.; Dalgleish, A.; Lasky, L.; Berman, P. Variable and conserved neutralization antigens of human immunodeficiency virus. Nature 1986, 324, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Wrin, T.; Little, S.J.; Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 2002, 100, 4144–4149. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Seaman, M.S.; Velinzon, K.; Pietzsch, J.; Ott, R.G.; Anthony, R.M.; Zebroski, H.; Hurley, A.; et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009, 458, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Bar, K.J.; Tsao, C.-y.; Iyer, S.S.; Decker, J.M.; Yang, Y.; Bonsignori, M.; Chen, X.; Hwang, K.-K.; Montefiori, D.C.; Liao, H.-X.; et al. Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog. 2012, 8, e1002721. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Bonsignori, M.; Liao, H.X.; Kumar, A.; Xia, S.M.; Lu, X.; Cai, F.; Hwang, K.K.; Song, H.; Zhou, T.; et al. Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 2014, 158, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Hraber, P.; Seaman, M.S.; Bailer, R.T.; Mascola, J.R.; Montefiori, D.C.; Korber, B.T. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 2014, 28, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, N.; Liu, M.; Salazar-Gonzalez, J.; Ferrari, G.; Giorgi, E.; Ganusov, V.; Keele, B.; Learn, G.; Turnbull, E.; Salazar, M.; et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009, 206, 1253–1272. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE 2010, 5, e12303. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.K.; Hawkins, N.; Ritchie, A.J.; Ganusov, V.V.; Whale, V.; Brackenridge, S.; Li, H.; Pavlicek, J.W.; Cai, F.; Rose-Abrahams, M.; et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J. Clin. Invest. 2013, 123, 380–393. [Google Scholar] [PubMed]
- Frost, S.D.; Wrin, T.; Smith, D.M.; Kosakovsky Pond, S.L.; Liu, Y.; Paxinos, E.; Chappey, C.; Galovich, J.; Beauchaine, J.; Petropoulos, C.J.; et al. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl. Acad. Sci. USA 2005, 102, 18514–18519. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.T.; Holmes, E.C.; Pybus, O.G.; Wilson, D.J.; Viscidi, R.P.; Abrams, E.J.; Phillips, R.E.; Drummond, A.J. Evolution of the human immunodeficiency virus envelope gene is dominated by purifying selection. Genetics 2006, 174, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.; Kosakovsky Pond, S.L.; Bennett, P.; Richman, D.D.; Leigh Brown, A.J.; Frost, S.D. Adaptation to human populations is revealed by within-host polymorphisms in HIV-1 and hepatitis C virus. PLoS Pathog. 2007, 3, e45. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Z.Y.; Li, Y.; Hogerkorp, C.M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.P.; Wang, S.K.; Ramos, A.; Chan-Hui, P.Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Mouquet, H.; Ueberheide, B.; Diskin, R.; Klein, F.; Oliveira, T.Y.; Pietzsch, J.; Fenyo, D.; Abadir, A.; Velinzon, K.; et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 2011, 333, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Kepler, T.B. Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res 2013, 2, 103. [Google Scholar] [CrossRef] [PubMed]
- Kepler, T.B.; Munshaw, S.; Wiehe, K.; Zhang, R.; Yu, J.S.; Woods, C.W.; Denny, T.N.; Tomaras, G.D.; Alam, S.M.; Moody, M.A.; et al. Reconstructing a B-cell clonal lineage. II. Mutation, selection, and affinity maturation. Front. Immunol. 2014, 5, e170. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, C.K.; Bhiman, J.N.; Gray, E.S.; Tumba, N.; Abdool Karim, S.S.; Williamson, C.; Morris, L.; Moore, P.L. Viral escape from HIV-1 neutralizing antibodies drives increased plasma neutralization breadth through sequential recognition of multiple epitopes and immunotypes. PLoS Pathog. 2013, 9, e1003738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, B.F.; McElrath, M.J. Progress in HIV-1 vaccine development. Curr. Opin. HIV AIDS 2013, 8, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Moody, M.A.; Alam, S.M.; Bonsignori, M.; Verkoczy, L.; Ferrari, G.; Gao, F.; Tomaras, G.D.; Liao, H.X.; Kelsoe, G. Progress in HIV-1 vaccine development. J. Allergy Clin. Immunol. 2014, 134, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Haynes, B.F. HIV-1-specific antibody responses during acute and chronic HIV-1 infection. Curr. Opin. HIV AIDS 2009, 4, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.M.; Haynes, B.F. HIV-1 neutralizing antibodies: Understanding nature’s pathways. Immunol. Rev. 2013, 254, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.; Mascola, J. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Kelsoe, G.; Harrison, S.C.; Kepler, T.B. B-cell–lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 2012, 30, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Z.; Schramm, C.A.; Joyce, M.G.; Do Kwon, Y.; Zhou, T.; Sheng, Z.; Zhang, B.; O'Dell, S.; McKee, K.; et al. Maturation and diversity of the VRC01-antibody lineage over 15 years of chronic HIV-1 infection. Cell 2015, 161, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Bonsignori, M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; Morris, L.; Gray, E.; Marshall, D.J.; Crump, J.A.; Kapiga, S.H.; Sam, N.E.; et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 2011, 85, 9998–10009. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Mascola, J.R. Human antibodies that neutralize HIV: Identification, structures, and B cell ontogenies. Immunity 2012, 37, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.A.; Gao, F.; Gurley, T.C.; Amos, J.D.; Kumar, A.; Hora, B.; Marshall, D.J.; Whitesides, J.F.; Xia, S.M.; Parks, R.; et al. Strain-specific V3 and CD4 binding site antibodies constrain autologous HIV-1 to a neutralization-resistant phenotype. Cell Host Microbe 2015, 18, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ganusov, V.; Goonetilleke, N.; Liu, M.; Ferrari, G.; Shaw, G.; McMichael, A.; Borrow, P.; Korber, B.; Perelson, A. Fitness costs and diversity of CTL response determine the rate of CTL escape during the acute and chronic phases of HIV infection. J. Virol. 2011, 85, 10518–10528. [Google Scholar] [CrossRef] [PubMed]
- Batorsky, R.; Sergeev, R.A.; Rouzine, I.M. The route of HIV escape from immune response targeting multiple sites is determined by the cost-benefit tradeoff of escape mutations. PLoS Comput. Biol. 2014, 10, e1003878. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Haubold, B.; Hauert, C.; Traulsen, A. Emergence of stable polymorphisms driven by evolutionary games between mutants. Nat. Commun. 2012, 3, 919. [Google Scholar] [PubMed]
- Korber, B.; Foley, B.; Leitner, T.; Kuiken, C.; Pillai, S.K.; Sodroski, J.G. Numbering positions in HIV relative to HXB2CG. In Human Retroviruses and AIDS 1998; Korber, B., Kuiken, C., Foley, B., Hahn, B.H., McCutchan, F., Mellors, J.W., Sodroski, J., Eds.; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 1998; pp. III–102–III–111. [Google Scholar]
- Wu, X.; Zhou, T.; Zhu, J.; Zhang, B.; Georgiev, I.; Wang, C.; Chen, X.; Longo, N.S.; Louder, M.; McKee, K.; et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 2011, 333, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Scharf, L.; Horwitz, J.; Klein, F.; Nussenzweig, M.C.; Bjorkman, P.J. Computational analysis of anti-HIV-1 antibody neutralization panel data to identify potential functional epitope residues. Proc. Natl. Acad. Sci. USA 2013, 110, 10598–10603. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Zhou, T.; Druz, A.; Georgiev, I.S.; Soto, C.; Gorman, J.; Huang, J.; Acharya, P.; Chuang, G.Y.; Ofek, G.; et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 2014, 514, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Diskin, R.; Scheid, J.F.; Marcovecchio, P.M.; West, A.P., Jr.; Klein, F.; Gao, H.; Gnanapragasam, P.N.; Abadir, A.; Seaman, M.S.; Nussenzweig, M.C.; et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 2011, 334, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van Ryk, D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007, 445, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Bonsignori, M.; Alam, S.M.; McLellan, J.S.; Tomaras, G.D.; Moody, M.A.; Kozink, D.M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity 2013, 38, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Fera, D.; Schmidt, A.G.; Haynes, B.F.; Gao, F.; Liao, H.X.; Kepler, T.B.; Harrison, S.C. Affinity maturation in an HIV broadly neutralizing B-cell lineage through reorientation of variable domains. Proc. Natl. Acad. Sci. USA 2014, 111, 10275–10280. [Google Scholar] [CrossRef] [PubMed]
- HIV genome browser. Available online: hiv.lanl.gov/content/sequence/genome_browser/ browser.html (accessed on 13 October 2015).
- CATNAP: Compile, analyze and tally NAb panels. Available online: hiv.lanl.gov/components/ sequence/HIV/neutralization/ (accessed on 13 October 2015).
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.L.; Gray, E.S.; Wibmer, C.K.; Bhiman, J.N.; Nonyane, M.; Sheward, D.J.; Hermanus, T.; Bajimaya, S.; Tumba, N.L.; Abrahams, M.R.; et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med. 2012, 18, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Lee, J.H.; Doores, K.J.; Murin, C.D.; Julien, J.P.; McBride, R.; Liu, Y.; Marozsan, A.; Cupo, A.; Klasse, P.J.; et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 2013, 20, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A. Clustering Algorithms; Wiley: New York, 1975; p. 351. [Google Scholar]
- Kaufman, L.; Rousseew, P.J. Finding groups in data: An introduction to cluster analysis; John Wiley and Sons: Hoboken, 2005; p. 342. [Google Scholar]
- Pennings, P.S.; Kryazhimskiy, S.; Wakeley, J. Loss and recovery of genetic diversity in adapting populations of HIV. PLoS Genet. 2014, 10, e1004000. [Google Scholar] [PubMed]
- Salazar-Gonzalez, J.; Salazar, M.; Keele, B.; Learn, G.; Giorgi, E.; Li, H.; Decker, J.; Wang, S.; Baalwa, J.; Kraus, M.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Bailes, E.; Pham, K.T.; Salazar, M.G.; Guffey, M.B.; Keele, B.F.; Derdeyn, C.A.; Farmer, P.; Hunter, E.; Allen, S.; et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 2008, 82, 3952–3970. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T.J.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012, 8, e1002529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, S.J.; Murray, J.M. The unrooted set covering connected subgraph problem differentiating between HIV envelope sequences. Eur. J. Oper. Res. 2016, 248, 668–680. [Google Scholar] [CrossRef]
- Murray, J.M.; Moenne-Loccoz, R.; Velay, A.; Habersetzer, F.; Doffoël, M.; Gut, J.P.; Fofana, I.; Zeisel, M.B.; Stoll-Keller, F.; Baumert, T.F.; et al. Genotype 1 hepatitis C virus envelope features that determine antiviral response assessed through optimal covariance networks. PLoS ONE 2013, 8, e67254. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Gnanakaran, S. The implications of patterns in HIV diversity for neutralizing antibody induction and susceptibility. Curr. Opin. HIV AIDS 2009, 4, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Sather, D.N.; Carbonetti, S.; Malherbe, D.; Pissani, F.; Stuart, A.B.; Hessell, A.J.; Gray, M.D.; Mikell, I.; Kalams, S.A.; Haigwood, N.L.; et al. Emergence of broadly neutralizing antibodies and viral co-evolution in two subjects during the early stages of infection with human immunodeficiency virus type 1. J. Virol. 2014, 88, 12968–12981. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mata-Fink, J.; Kriegsman, B.; Hanson, M.; Irvine, D.J.; Eisen, H.N.; Burton, D.R.; Wittrup, K.D.; Kardar, M.; Chakraborty, A.K. Manipulating the selection forces during affinity maturation to generate cross-reactive HIV antibodies. Cell 2015, 160, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, D.C.; Doria-Rose, N.A.; Misher, L.; Beckett, T.; Puryear, W.B.; Schuman, J.T.; Kraft, Z.; O'Malley, J.; Mori, M.; Srivastava, I.; et al. Sequential immunization with a subtype B HIV-1 envelope quasispecies partially mimics the in vivo development of neutralizing antibodies. J. Virol. 2011, 85, 5262–5274. [Google Scholar] [CrossRef] [PubMed]
- Pissani, F.; Malherbe, D.C.; Robins, H.; DeFilippis, V.R.; Park, B.; Sellhorn, G.s.; Stamatatos, L.; Overbaugh, J.; Haigwood, N.L. Motif-optimized subtype A HIV envelope-based DNA vaccines rapidly elicit neutralizing antibodies when delivered sequentially. Vaccine 2012, 30, 5519–5526. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, D.C.; Pissani, F.; Sather, D.N.; Guo, B.; Pandey, S.; Sutton, W.F.; Stuart, A.B.; Robins, H.; Park, B.; Krebs, S.J.; et al. Envelope variants circulating as initial neutralization breadth developed in two HIV-infected subjects stimulate multiclade neutralizing antibodies in rabbits. J. Virol. 2014, 88, 12949–12967. [Google Scholar] [CrossRef] [PubMed]
- McIlroy, D.; Barteau, B.; Cany, J.; Richard, P.; Gourden, C.; Conchon, S.; Pitard, B. DNA/amphiphilic block co-polymer nanospheres promote low-dose DNA vaccination. Mol. Ther. 2009, 17, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Chèvre, R.; Le Bihan, O.; Beilvert, F.; Chatin, B.; Barteau, B.; Mével, M.; Lambert, O.; Pitard, B. Amphiphilic block copolymers enhance the cellular uptake of DNA molecules through a facilitated plasma membrane transport. Nucleic Acids Res. 2011, 39, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lewis-Ximenez, L.L.; Jones, A.; Nolan, B.; Schulze zur Wiesch, J.; Li, B.; Schneidewind, A.; Kim, A.Y.; et al. Viral sequence evolution in acute hepatitis C virus infection. J. Virol. 2007, 81, 11658–11668. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.; Netski, D.; Wang, X.; Cox, A.; Ray, S. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 2009, 136, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fisher, B.; Dowd, K.; Astemborski, J.; Cox, A.; Ray, S. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J. Virol. 2010, 84, 5067–5077. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Stoddard, M.B.; Wang, S.; Blair, L.M.; Giorgi, E.E.; Parrish, E.H.; Learn, G.H.; Hraber, P.; Goepfert, P.A.; Saag, M.S.; et al. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS Pathog. 2012, 8, e1002880. [Google Scholar] [CrossRef] [PubMed]
- Tarr, A.W.; Khera, T.; Hueging, K.; Sheldon, J.; Steinmann, E.; Pietschmann, T.; Brown, R.J. Genetic diversity underlying the envelope glycoproteins of hepatitis C virus: Structural and functional consequences and the implications for vaccine design. Viruses 2015, 7, 3995–4046. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [PubMed]
- Pond, S.L.; Frost, S.D.; Grossman, Z.; Gravenor, M.B.; Richman, D.D.; Brown, A.J. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2006, 2, e62. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Multiple alignment using hidden Markov models. ISMB 1995, 3, 114–120. [Google Scholar] [PubMed]
- Gaschen, B.; Kuiken, C.; Korber, B.; Foley, B. Retrieval and on-the-fly alignment of sequence fragments from the HIV database. Bioinformatics 2001, 17, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Nickle, D.C.; Heath, L.; Jensen, M.A.; Gilbert, P.B.; Mullins, J.I.; Kosakovsky-Pond, S.L. HIV-specific probabilistic models of protein evolution. PLoS ONE 2007, 2, e503. [Google Scholar] [CrossRef] [PubMed]
- Nemhauser, G.L.; Wolsey, L.A. Maximizing submodular functions: Formulations and analysis of algorithms. Ann. Discrete Math. 1981, 11, 279–301. [Google Scholar]
- LASSIE repository. Available online: www.github.com/phraber/lassie (accessed on 13 October 2015).
- Fischer, W. About the cover. In HIV Sequence Compendium 2011; Kuiken, C., Foley, B., Leitner, T., Apetrei, C., Hahn, B.H., Mizrachi, I., Mullins, J.I., Rambaut, A., Wolinsky, S.M., Korber, B., Eds.; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 2011; pp. 2–3. [Google Scholar]
- Brodin, J.; Krishnamoorthy, M.; Athreya, G.; Fischer, W.; Hraber, P.; Gleasner, C.; Green, L.; Korber, B.; Leitner, T. A multiple-alignment based primer design algorithm for genetically highly variable DNA targets. BMC Bioinformatics 2013, 14, 255. [Google Scholar] [CrossRef] [PubMed]
- PixelGram repository. Available online: www.github.com/phraber/pixelgram (accessed on 13 October 2015).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Lanave, C.; Preparata, G.; Saccone, C.; Serio, G. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 1984, 20, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hraber, P.; Korber, B.; Wagh, K.; Giorgi, E.E.; Bhattacharya, T.; Gnanakaran, S.; Lapedes, A.S.; Learn, G.H.; Kreider, E.F.; Li, Y.; et al. Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants. Viruses 2015, 7, 5443-5475. https://doi.org/10.3390/v7102881
Hraber P, Korber B, Wagh K, Giorgi EE, Bhattacharya T, Gnanakaran S, Lapedes AS, Learn GH, Kreider EF, Li Y, et al. Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants. Viruses. 2015; 7(10):5443-5475. https://doi.org/10.3390/v7102881
Chicago/Turabian StyleHraber, Peter, Bette Korber, Kshitij Wagh, Elena E. Giorgi, Tanmoy Bhattacharya, S. Gnanakaran, Alan S. Lapedes, Gerald H. Learn, Edward F. Kreider, Yingying Li, and et al. 2015. "Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants" Viruses 7, no. 10: 5443-5475. https://doi.org/10.3390/v7102881
APA StyleHraber, P., Korber, B., Wagh, K., Giorgi, E. E., Bhattacharya, T., Gnanakaran, S., Lapedes, A. S., Learn, G. H., Kreider, E. F., Li, Y., Shaw, G. M., Hahn, B. H., Montefiori, D. C., Alam, S. M., Bonsignori, M., Moody, M. A., Liao, H. -X., Gao, F., & Haynes, B. F. (2015). Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants. Viruses, 7(10), 5443-5475. https://doi.org/10.3390/v7102881