
Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry)

 ,
,
Abstract
:1. Introduction
2. Results
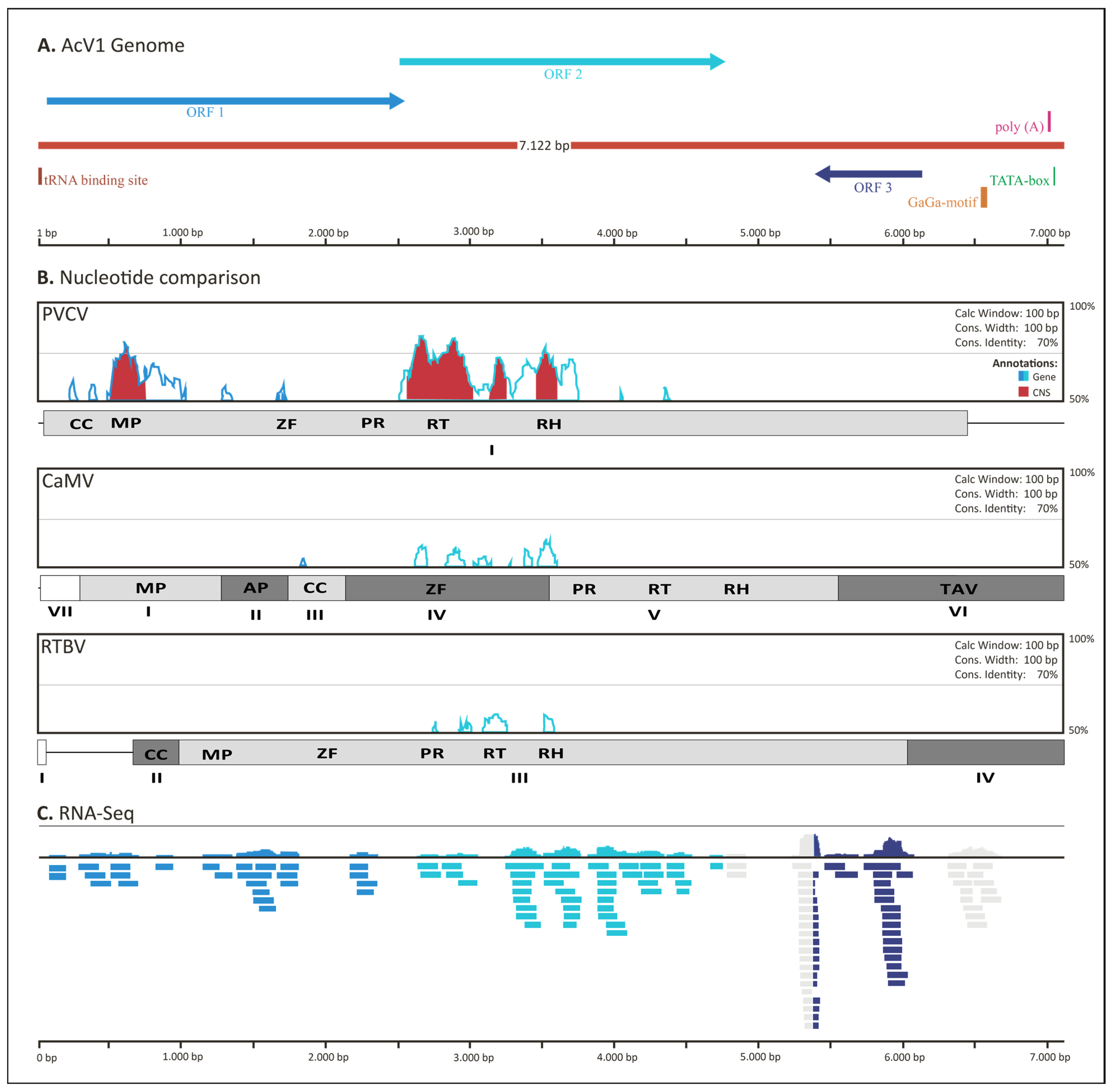
2.1. Deep Sequencing of Aristotelia Chilensis Genome Results in the Discovery of AcV1
2.2. Bioinformatics Characterization of AcV1

{kind=link}
{kind=link}
{kind=link}
| ORF Number | Start Position | End Position | Length (bp) | Predicted Length (aa) | Protein Weight (kDa) |
|---|---|---|---|---|---|
| 1 | 81 | 2543 | 2463 | 820 | 92.22 |
| 2 | 2540 | 4816 | 2277 | 758 | 88.69 |
| 3 | 6208 | 5408 | 800 | 266 | 29.91 |

2.3. Phylogenetic Analysis Reveals a Strong Relationship between AcV1 and the Caulimoviridae Family

2.4. General Genomic Organization of AcV1

2.5. Evidence of AcV1 Transcriptional Activity Based on Expression Levels Estimated by RNA-seq
3. Discussion
4. Materials and Methods
4.1. DNA Sequencing and Data Processing
4.2. Sequence Analysis and Genome Comparison
4.3. RNA Sequencing and Data Processing
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hansen, C.; Heslop-Harrison, J. Sequences and phylogenies of plant pararetroviruses, viruses, and transposable elements. Adv. Bot. Res. 2004, 41, 165–193. [Google Scholar]
- Barba, M.; Czosnek, H.; Hadidi, A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 2014, 6, 106–136. [Google Scholar] [CrossRef] [PubMed]
- Prabha, K.; Baranwal, V.K.; Jain, R.K. Applications of next generation high throughput sequencing technologies in characterization, discovery and molecular interaction of plant viruses. Indian J. Virol. 2013, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Araya, H.; Clavijo, C.; Herrera, C. Capacidad antioxidante de frutas y verduras cultivados en Chile. Arch. Latinoam. Nutr. 2006, 56, 361–365. (In Spanish) [Google Scholar] [PubMed]
- Escribano-bailón, M.T.; Alcalde-eon, C.; Muñoz, O.; Rivas-gonzalo, J.C.; Santos-buelga, C. Anthocyanins in Berries of Maqui [Aristotelia chilensis (Mol.) Stuntz]. Phytochem. Anal. 2006, 17, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Schreckinger, M.; Wang, J. Antioxidant capacity and in vitro inhibition of adipogenesis and inflammation by phenolic extracts of Vaccinium floribundum and Aristotelia chilensis. J. Agric. Food Chem. 2010, 58, 8966–8976. [Google Scholar] [CrossRef] [PubMed]
- Céspedes, C.L.; El-Hafidi, M.; Pavon, N.; Alarcon, J. Antioxidant and cardioprotective activities of phenolic extracts from fruits of Chilean blackberry Aristotelia chilensis (Elaeocarpaceae), Maqui. Food Chem. 2008, 107, 820–829. [Google Scholar] [CrossRef]
- Miranda-Rottmann, S.; Aspillaga, A.A.; Pérez, D.D.; Vasquez, L.; Martinez, A.L.F.; Leighton, F. Juice and phenolic fractions of the berry Aristotelia chilensis inhibit LDL oxidation in vitro and protect human endothelial cells against oxidative stress. J. Agric. Food Chem. 2002, 50, 7542–7547. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.; Hull, R.; Lockhart, B.; Olszewski, N. Viral sequences integrated into plant genomes. Annu. Rev. Phytopathol. 2002, 40, 119–136. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Hohn, T. Involvement of reverse transcription in the replication of cauliflower mosaic virus: A detailed model and test of some aspects. Cell 1983, 33, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J. Artemis: Sequence visualization and annotation. Bioinformatics (Oxf. Engl.) 2000, 16, 944–945. [Google Scholar] [CrossRef]
- Hull, R.; Sadler, J.; Longstaff, M. The sequence of carnation etched ring virus DNA: Comparison with cauliflower mosaic virus and retroviruses. EMBO J. 1986, 5, 3083–3090. [Google Scholar] [PubMed]
- Hagen, L.; Jacquemond, M.; Lepingle, A.; Lot, H.; Tepfer, M. Nucleotide sequence and genomic organization of cacao swollen shoot virus. Virology 1993, 196, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.M.; Jones, M.C.; Blakebrough, M.L.; Dasgupta, I.; Davies, J.W.; Hull, R. An analysis of the sequence of an infectious clone of rice tungro bacilliform virus, a plant pararetrovirus. Nucleic Acids Res. 1991, 19, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Richert-Pöggeler, K.R.; Shepherd, R.J. Petunia vein-clearing virus: A plant pararetrovirus with the core sequences for an integrase function. Virology 1997, 236, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Shao, J.; Schneider, W.L.; Hartung, J.S.; Brlansky, R.H. Population of endogenous pararetrovirus genomes in carrizo citrange. Genome Announc. 2014. [Google Scholar] [CrossRef]
- Richins, R. Sequence of figwort mosaic virus DNA (caulimovirus group). Nucleic Acids Res. 1987, 15, 8451–8466. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. 2011, 7, 539. [Google Scholar] [CrossRef]
- Koonin, E.V.; Mushegian, A.R.; Ryabov, E.V.; Dolja, V.V. Diverse groups of plant RNA and DNA viruses share related movement proteins that may possess chaperone-like activity. J. Gen. Virol. 1991, 72, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Kasteel, D.T.; Perbal, M.C.; Boyer, J.C.; Wellink, J.; Goldbach, R.W.; Maule, A.J.; van Lent, J.W. The movement proteins of cowpea mosaic virus and cauliflower mosaic virus induce tubular structures in plant and insect cells. J. Gen. Virol. 1996, 77, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Piqué, M.; Mougeot, J.L.; Geldreich, A.; Guidasci, T.; Mesnard, J.M.; Lebeurier, G.; Yot, P. Sequence of a cauliflower mosaic virus strain infecting solanaceous plants. Gene 1995, 155, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Eickbush, T. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362. [Google Scholar] [PubMed]
- Hansen, L.; Sandmeyer, S. Characterization of a transpositionally active Ty3 element and identification of the Ty3 integrase protein. J. Virol. 1990, 64, 2599–2607. [Google Scholar] [PubMed]
- Malik, H.; Eickbush, T. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001, 11, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Kano, H.; Koizumi, M.; Noda, H.; Hibino, H. Nucleotide sequence of capsid protein gene of rice tungro bacilliform virus. Arch. Virol. 1992, 124, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Almeyda, C.V.; Eid, S.G.; Saar, D.; Samuitiene, M.; Pappu, H.R. Comparative analysis of endogenous plant pararetroviruses in cultivated and wild Dahlia spp. Virus Genes 2014, 48, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Maumus, F.; Copetti, D.; Choisne, N.; Zwickl, D.J.; Zytnicki, M.; McTaggart, A.R.; Scalabrin, S.; Vezzulli, S.; Wing, R.A.; et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat. Commun. 2014, 5, 5269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.; Schwartz, R. A model of evolutionary change in proteins. Atlas Protein Seq. Struct. 1978, 5, 345–351. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Calvert, L.A.; Ospina, M.D.; Shepherd, R.J. Characterization of cassava vein mosaic virus: A distinct plant pararetrovirus. J. Gen. 1995, 76, 1271–1276. [Google Scholar]
- Medberry, S. Properties of Commelina yellow mottle virus’s complete DNA sequence, genomic discontinuities and transcript suggest that it is a pararetrovirus. Nucleic Acids 1990, 18, 5505–5513. [Google Scholar] [CrossRef]
- He, X.; Fütterer, J.; Hohn, T. Contribution of downstream promoter elements to transcriptional regulation of the rice tungro bacilliform virus promoter. Nucleic Acids Res. 2002, 30, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Gayral, P.; Noa-Carrazana, J.C.; Lescot, M.; Lheureux, F.; Lockhart, B.E.L.; Matsumoto, T.; Piffanelli, P.; Iskra-Caruana, M.L. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J. Virol. 2008, 82, 6697–6710. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, B.E.; Menke, J.; Dahal, G.; Olszewski, N.E. Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 2000, 81, 1579–1585. [Google Scholar] [PubMed]
- Richert-Pöggeler, K.R.; Noreen, F.; Schwarzacher, T.; Harper, G.; Hohn, T. Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 2003, 22, 4836–4845. [Google Scholar] [CrossRef] [PubMed]
- Noreen, F.; Akbergenov, R.; Hohn, T.; Richert-Pöggeler, K.R. Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J. Cell Mol. Biol. 2007, 50, 219–229. [Google Scholar] [CrossRef]
- Chabannes, M.; Iskra-Caruana, M.L. Endogenous pararetroviruses-a reservoir of virus infection in plants. Curr. Opin. Virol. 2013, 3, 8–13. [Google Scholar] [CrossRef]
- Staginnus, C.; Richert-Pöggeler, K.R. Endogenous pararetroviruses: Two-faced travelers in the plant genome. Trends Plant Sci. 2006, 11, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Iskra-Caruana, M.L.; Lockhart, B.; Hohn, T.; Richert-Pöggeler, K.R. Suggestions for a nomenclature of endogenous pararetroviral sequences in plants. Arch. Virol. 2009, 154, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Beld, M.; Martin, C.; Huits, H.; Stuitje, A.R.; Gerats, A.G. Flavonoid synthesis in Petunia hybrida: Partial characterization of dihydroflavonol-4-reductase genes. Plant Mol. Biol. 1989, 13, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Villacreses, J.; Rojas-Herrera, M.; Sanchez, C.; Blanc, N.; Espinoza, L.; Pastor, G.; Hewstone, N.; Undurraga, S.F.; Alzate, J.F.; Manque, P.; Maracaja-Coutinho, V.; Polanco, V. Draft genome sequence of the antioxidant-rich plant Maqui Berry (Aristotelia chilensis). (In preparation)
- Lodhi, M.; Ye, G.; Weeden, N.; Reisch, B. A simple and efficient method for DNA extraction from grapevine cultivars and Vitis species. Plant Mol. Biol. 1994, 12, 6–13. [Google Scholar] [CrossRef]
- Chevreux, B.; Pfisterer, T.; Drescher, B. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome 2004, 14, 1147–1159. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2 a multiple sequence alignment editor and analysis workbench. Bioinformatics (Oxf. Engl.) 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villacreses, J.; Rojas-Herrera, M.; Sánchez, C.; Hewstone, N.; Undurraga, S.F.; Alzate, J.F.; Manque, P.; Maracaja-Coutinho, V.; Polanco, V. Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry). Viruses 2015, 7, 1685-1699. https://doi.org/10.3390/v7041685
Villacreses J, Rojas-Herrera M, Sánchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V. Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry). Viruses. 2015; 7(4):1685-1699. https://doi.org/10.3390/v7041685
Chicago/Turabian StyleVillacreses, Javier, Marcelo Rojas-Herrera, Carolina Sánchez, Nicole Hewstone, Soledad F. Undurraga, Juan F. Alzate, Patricio Manque, Vinicius Maracaja-Coutinho, and Victor Polanco. 2015. "Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry)" Viruses 7, no. 4: 1685-1699. https://doi.org/10.3390/v7041685
APA StyleVillacreses, J., Rojas-Herrera, M., Sánchez, C., Hewstone, N., Undurraga, S. F., Alzate, J. F., Manque, P., Maracaja-Coutinho, V., & Polanco, V. (2015). Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry). Viruses, 7(4), 1685-1699. https://doi.org/10.3390/v7041685