
Humanized-VHH Transbodies that Inhibit HCV Protease and Replication

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
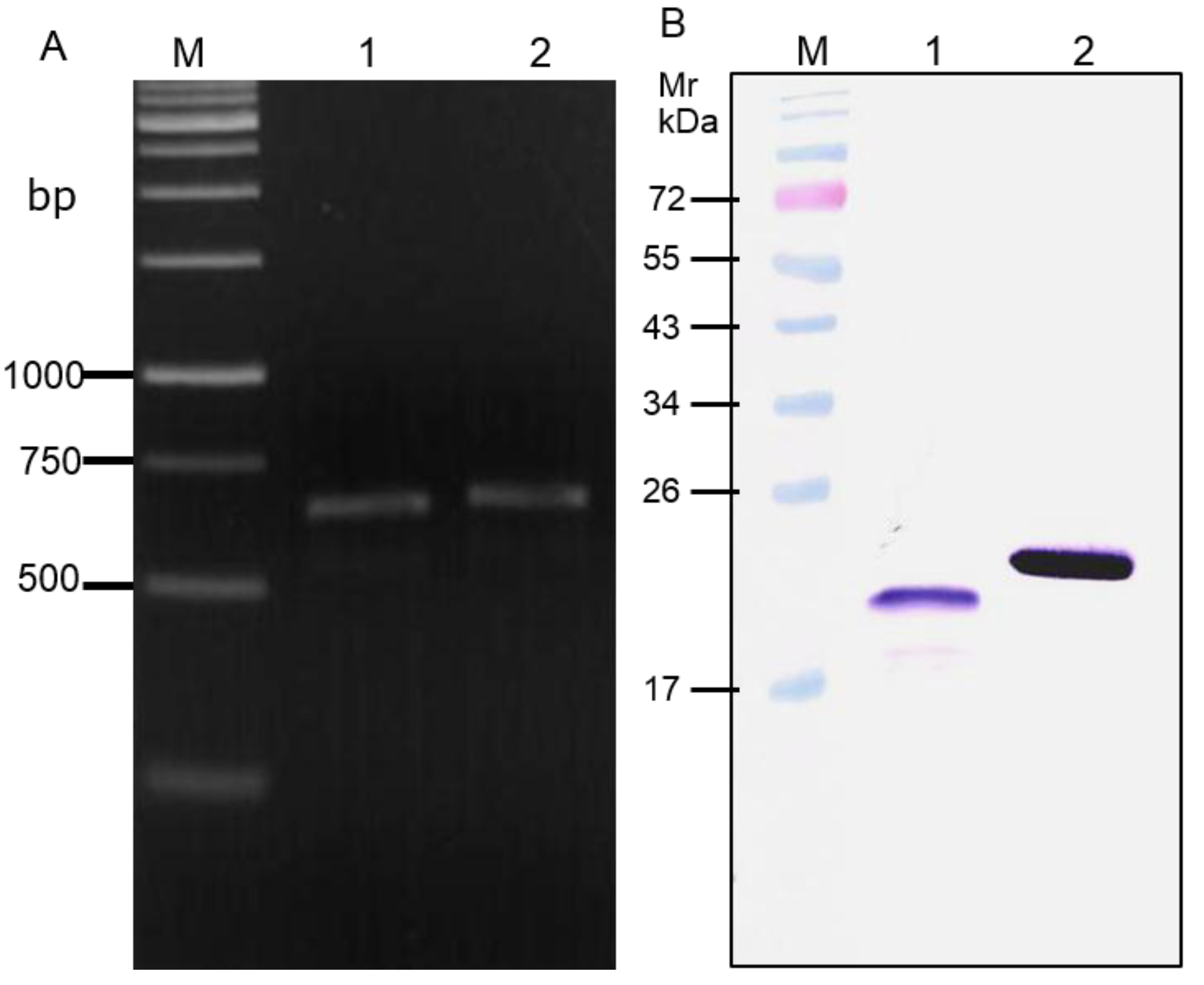
2.1. Production of Recombinant HCV NS3 and NS4A Fusion Protein (rNS3/4A)
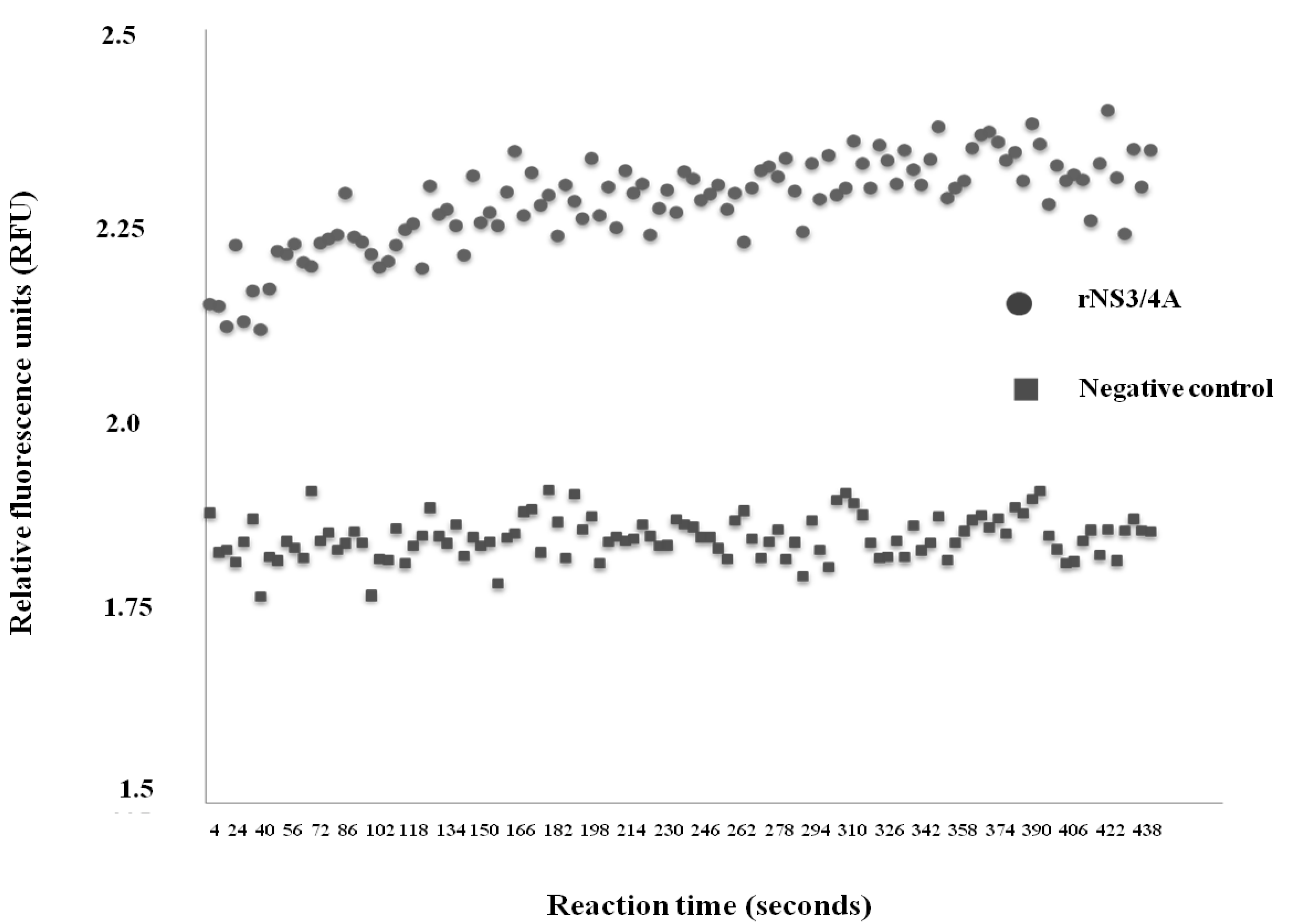
2.2. Determination of Protease Activity of the rNS3/4A
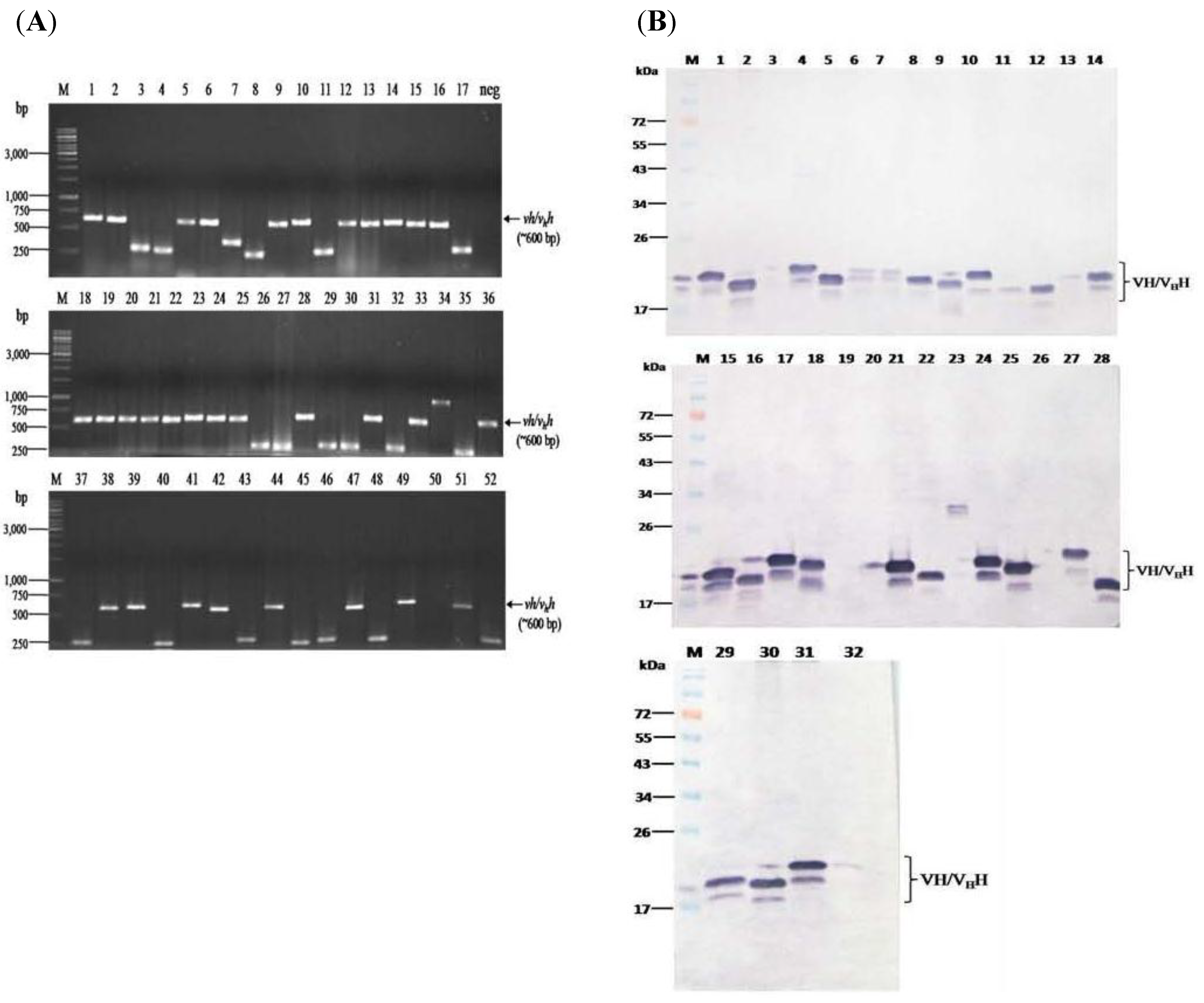
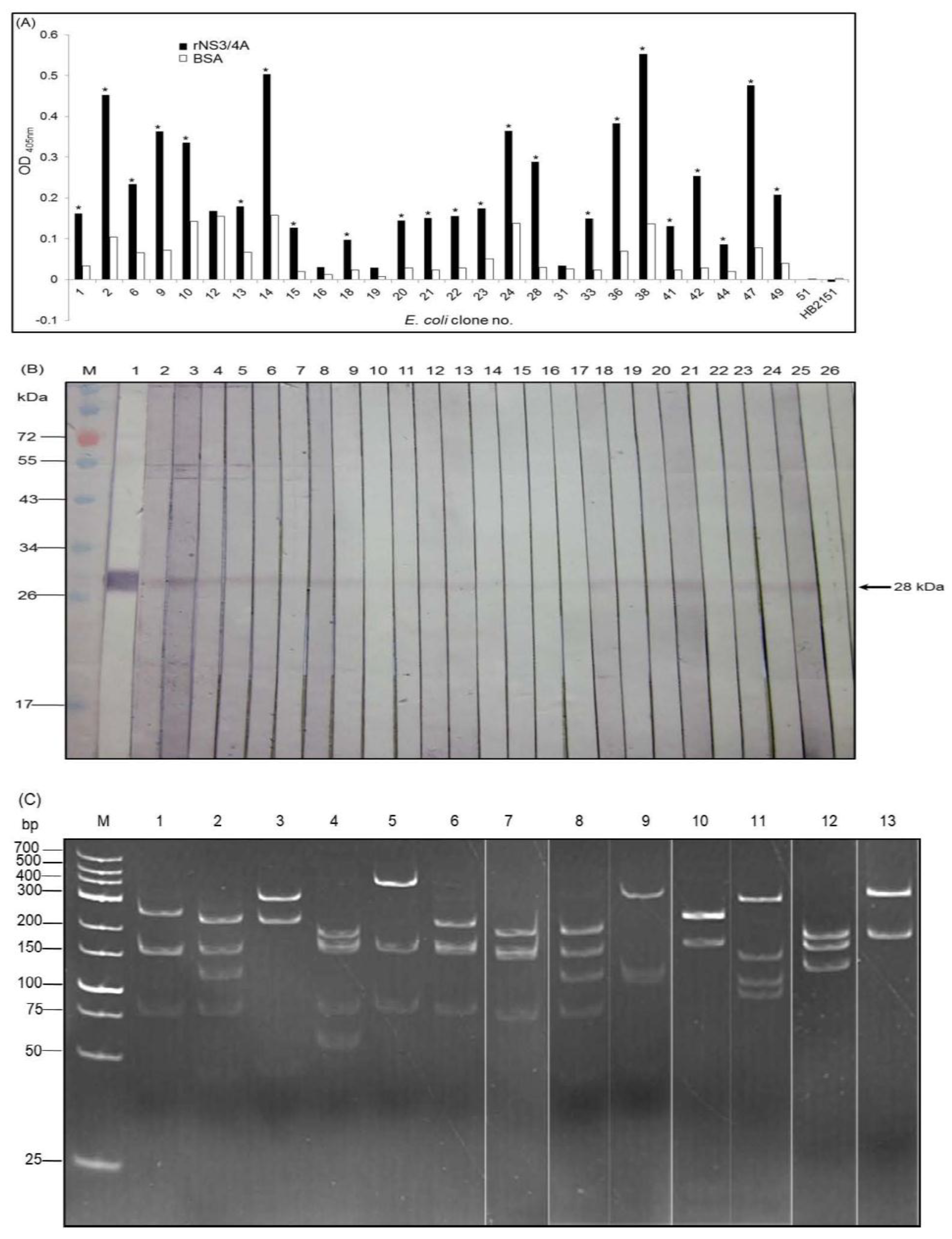
2.3. Production and Characterization of Soluble Nanobodies (VHs/VHHs) that Bound to rNS3/4A
2.4. Production of Cell-Penetrable Nanobodies (Transbodies)
2.5. Cell Penetrating Ability of the PEN-VHs/VHHs
2.6. LDH Assay
2.7. Inhibition of HCV Replication by HCV Protease Specific Cell Penetrable Nanobodies
2.8. Quantitative Real-Time RT-PCR (qRT-PCR)
2.9. Foci Assay
2.10. ELISA for Quantification of HCV Core Antigen
2.11. Response of the HCV RNA Transfected Cells to Treatment with Protease Specific-Cell Penetrable Nanobodies
2.12. Molecular Docking to Determine the Interaction between the Nanobodies and the HCV NS3/4A
2.13. Statistical Analysis
3. Results
3.1. Recombinant NS3/4A Protease

3.2. HCV Protease Specific Nanobodies and Their Characteristics


3.3. Cell Penetrable VHHs

3.4. Cellular Internalization and Cytotoxicity of the PEN-VHHs

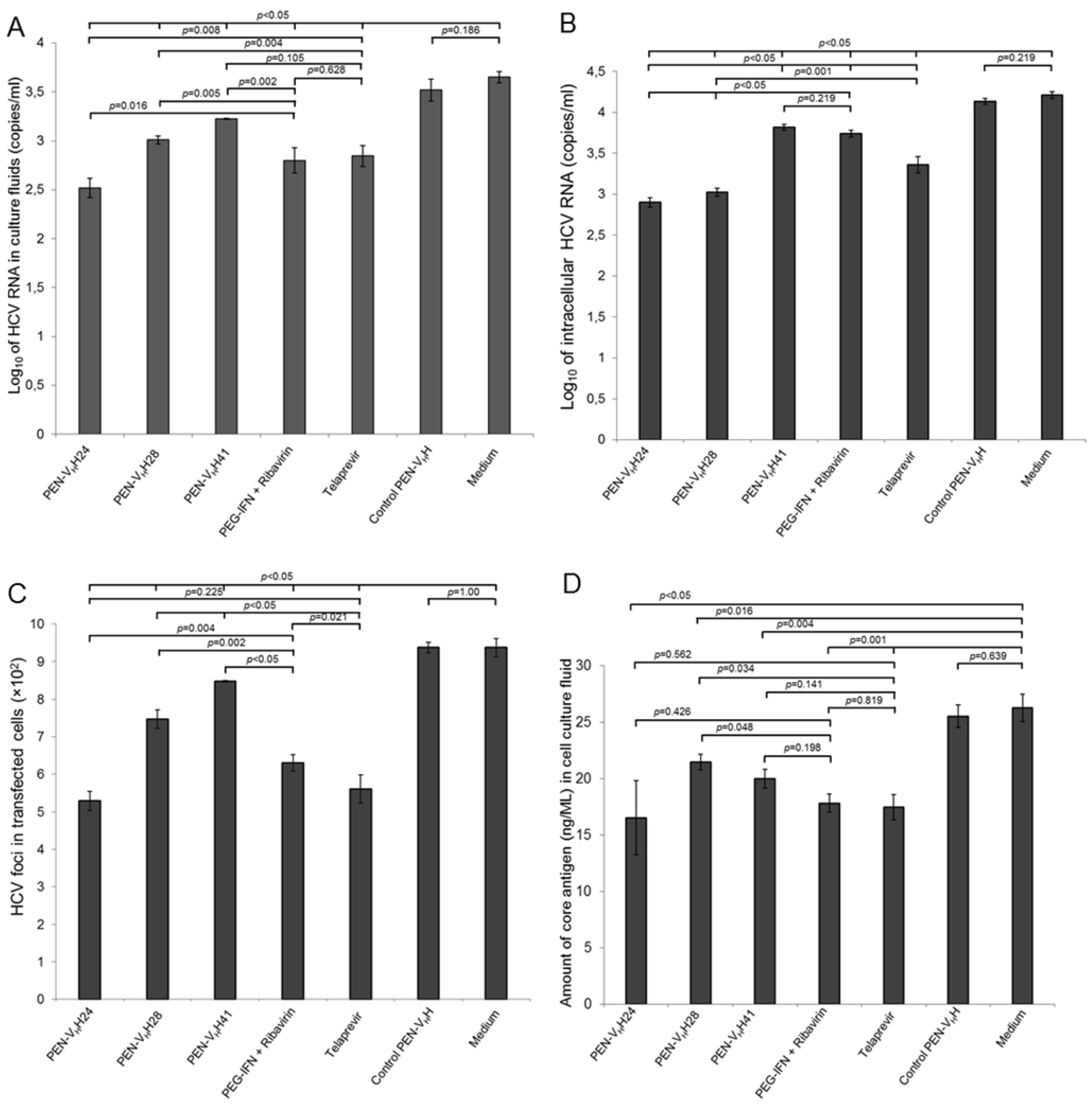
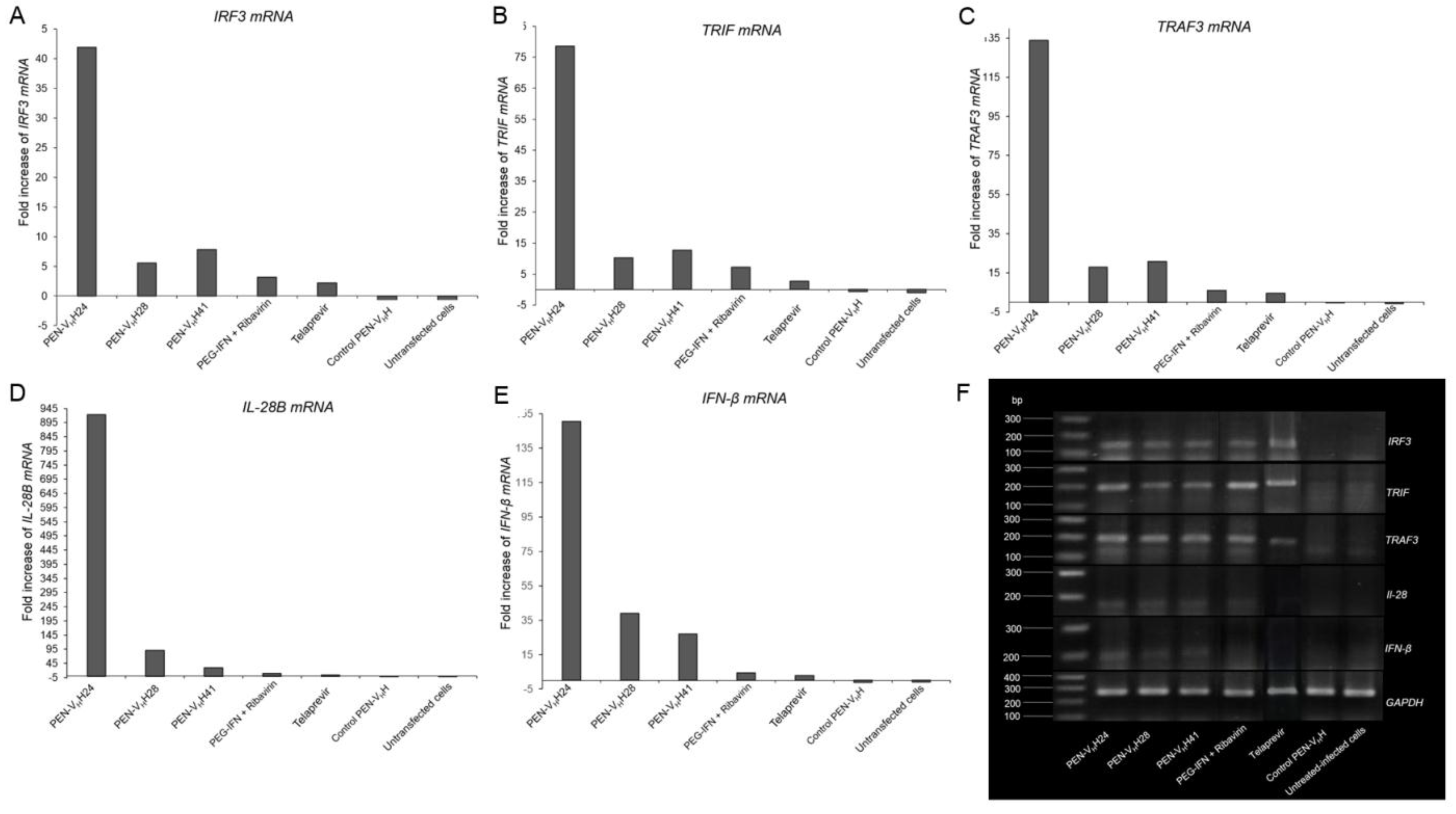
3.5. Inhibition of the HCV Replication by the Protease Specific-Transbodies


3.6. Response of the HCV Transfected-Cells to Treatment with Protease Specific-Transbodies

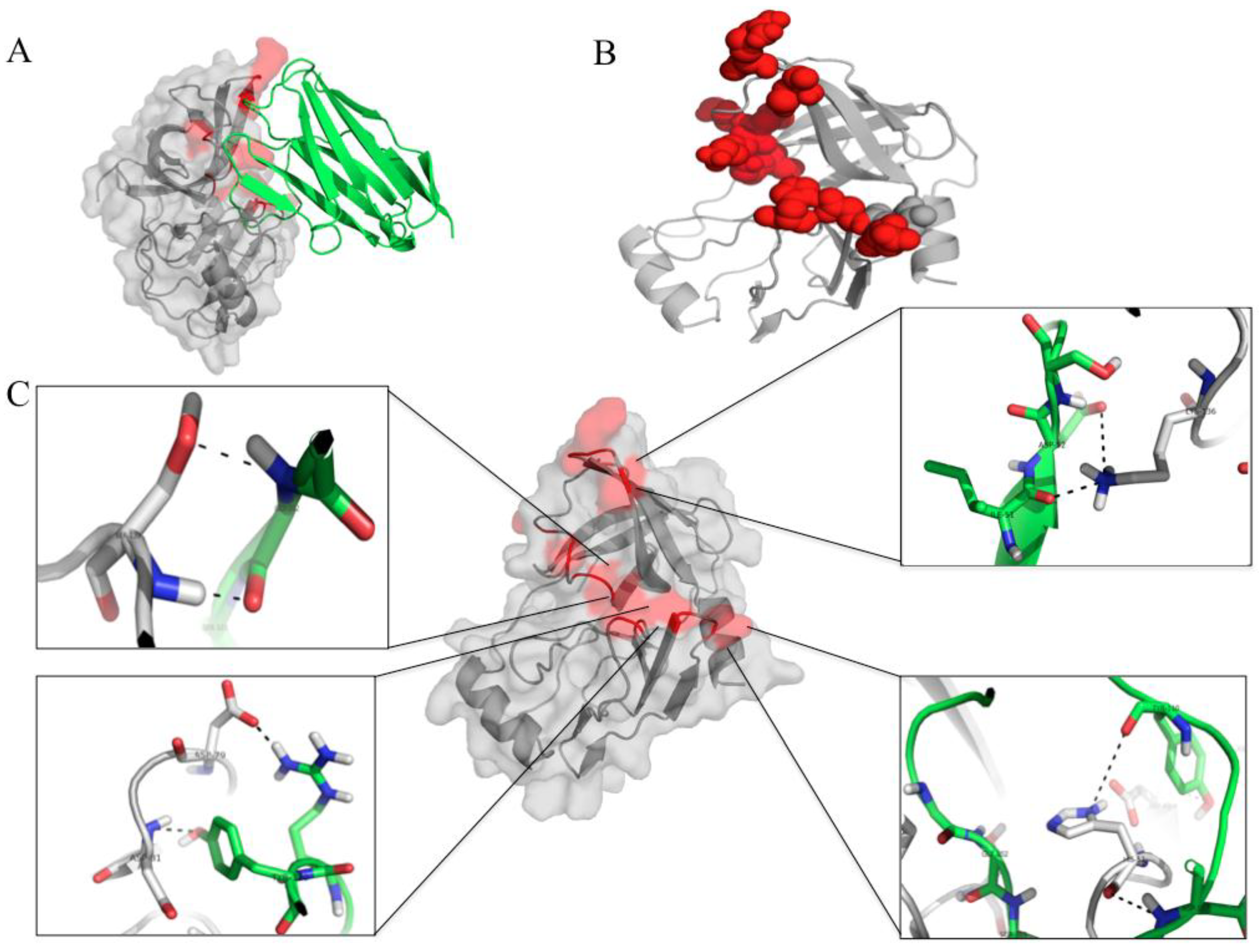
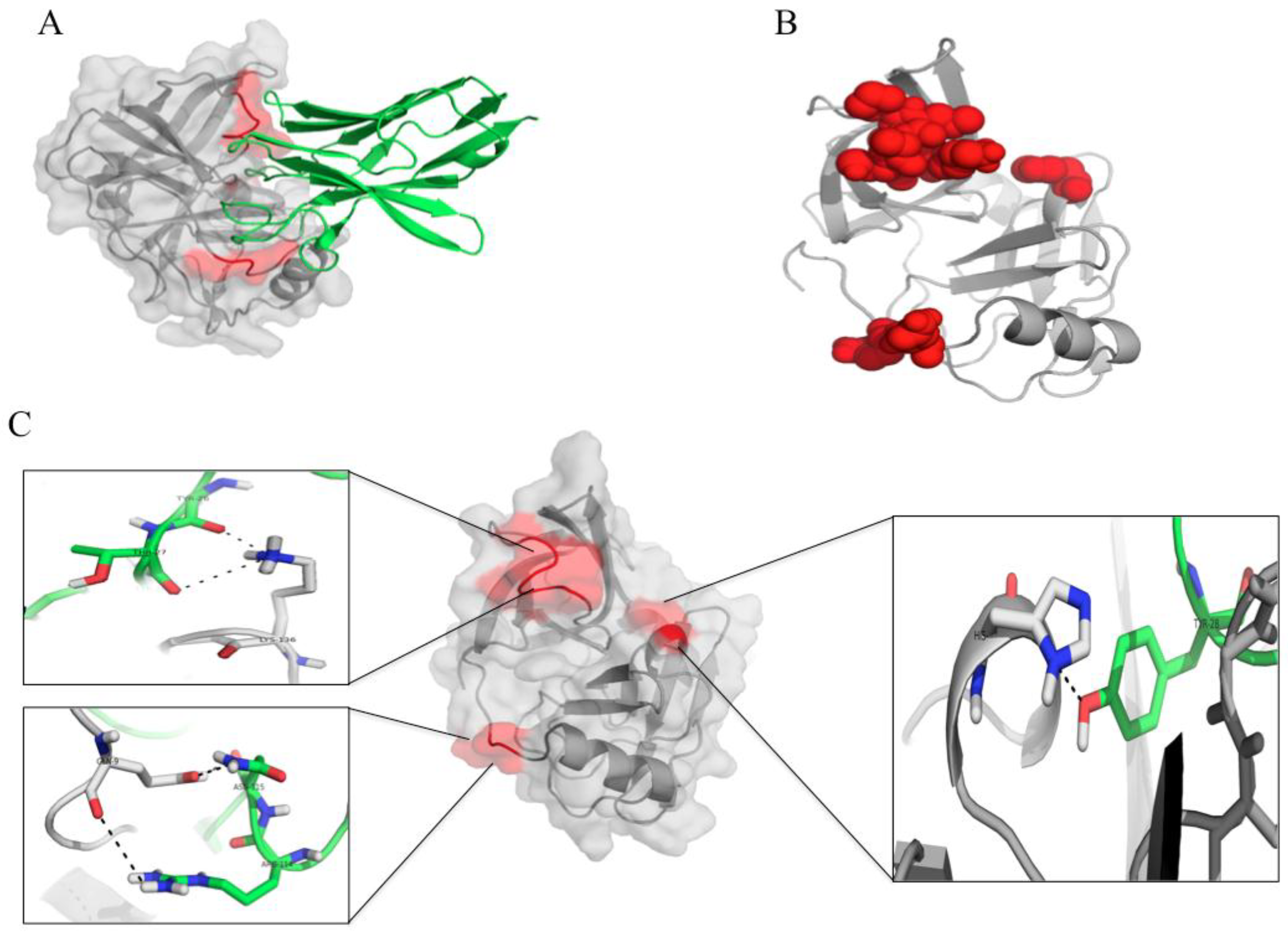
3.7. Computerized Binding of the Nanobodies to the HCV NS3/4A Protease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCV Protein | Humanized-VHH24 | Intermolecular Bond | |||
|---|---|---|---|---|---|
| NS3 Residue | Polyprotein Residue | Motif | Amino Acid | Domain | |
| Y56 | Y1088 | α -helix a | D107 | CDR3 | H bond |
| H57 | H1089 | α-helix a (catalytic triad) | Y110 | CDR3 | H bond |
| D81 | D1113 | Between β-E1 and β-F1 (catalytic triad) | Y110 | CDR3 | H-bond |
| A131 | A1163 | α-helix b | I51 | CDR2 | Hydrophobic |
| K136 | K1168 | Near α-helix b (oxyanion loop) | D50 | CDR2 | H bond |
| HCV Protein | Humanized-VHH28 | Intermolecular Bond | |||
|---|---|---|---|---|---|
| NS3 Residue | Polyprotein Residue | Motif | Amino Acid | Domain | |
| H57 | H1089 | α-helix a (catalytic triad) | D101 | CDR3 | H-bond |
| S61 | S1093 | Between α-helix a and β-E1 | D54 | CDR2 | H-bond |
| D81 | D1113 | Between α-helix a and β-E1 | Y102 | CDR3 | H-bond |
| N123 | N1155 | β-C2 | T73 | FR2 | H bond |
| L135 | L1167 | S1 pocket | W103 | CDR3 | Hydrohobic |
| HCV protein | Humanized-VHH41 | Intermolecular Bond | |||
|---|---|---|---|---|---|
| NS3 Residue | Polyprotein Residue | Motif | Amino Acid | Domain | |
| Q9 | Q1041 | Before α-helix a | N115 | CDR3 | H-bond |
| R11 | R1043 | α-helix a | R114 | CDR3 | Salt bridge |
| L13 | L1045 | α-helix a | W119 | FR4 | Hydrophobic |
| P131 | P1163 | α-helix b | N73 | FR3 | H bond |
| L135 | L1167 | S1 pocket | Q3 | FR1 | H bond |
| K136 | K1168 | Near α-helix b (oxyanion loop) | Y26 | CDR1 | H-bond |

4. Discussion
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interests
References
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peg-interferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Treatment Options for Patients with Chronic Hepatitis C not Responding to Initial Antiviral Therapy. Available online: http://vivo.med.cornell.edu/display/pubid69249101721 (accessed on 26 May 2014).
- Gelman, M.A.; Glenn, J.S. Mixing the right hepatitis C inhibitor cocktail. Trends Mol. Med. 2011, 17, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.; Chong, P.Y.; Shotwell, J.B.; Catalano, J.G.; Tai, V.W.; Fang, J.; Banka, A.L.; Roberts, C.D.; Youngman, M.; Zhang, H.; et al. Hepatitis C replication inhibitors that target the viral NS4B protein. J. Med. Chem. 2014, 57, 2107–2120. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.V.; Chung, R. Recent FDA approval of sofosbuvir and simeprevir. Implications for current HCV treatment. Clin. Liver Dis. 2014, 3, 65–68. [Google Scholar] [CrossRef]
- Wu, S.; Kanda, T.; Nakamoto, S.; Imazeki, F.; Yokosuka, O. Hepatitis C virus protease inhibitor-resistance mutations: Our experience and review. World J. Gastroenterol. 2013, 19, 8940–8948. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Kanwal, F. Boceprevir and telaprevir in the management of hepatitis C virus-infected patients. Clin. Infect. Dis. 2012, 54, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, F.Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Eng. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef]
- Stedman, C.A.M. Current prospects for interferon-free treatment of hepatitis C in 2012. J. Gastroenterol. Hepatol. 2013, 28, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ikeda, K.; Suzuki, F.; Toyota, J.; Karino, Y.; Chayama, K.; Kawakami, Y.; Ishikawa, H.; Watanabe, H.; Hu, W.; et al. Dual oral therapy with dalatasvia and asunaprevir for patients with HCV genotype 1b and limited treatment options. J. Hepatol. 2013, 58, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Rutter, G.; Kurpanek, K.; Bartenschlager, R. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 2001, 75, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 1994, 68, 5045–5055. [Google Scholar] [PubMed]
- Raney, K.D.; Sharma, S.D.; Moustafa, I.M.; Cameron, C.E. Hepatitis C virus non-structural protein 3 (HCV NS3): A multifunctional antiviral target. J. Biol. Chem. 2010, 285, 22725–22731. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Haidar, J.N.; Yuan, Q.-A.; Zeng, L.; Snavely, M.; Luna, X.; Zhang, H.; Zhu, W.; Ludwig, D.L.; Zhu, Z. A universal combinatorial design of antibody framework to graft distinct CDR sequences: A bioinformatics approach. Proteins 2012, 80, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frère, J.M.; Kinne, J.; Wyns, L.; Muyldermans, S. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteomics 2009, 72, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Thanongsaksrikul, J.; Srimanote, P.; Maneewatch, S.; Choowongkomon, K.; Tapchaisri, P.; Makino, S.; Kurazono, H.; Chaicumpa, W. A VHH that neutralizes the zinc metalloproteinase activity of botulinum neurotoxin type A. J. Biol. Chem. 2010, 285, 9657–9666. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Hamill, P.; Jean, F. Enzymatic characterization of membrane-associated hepatitis C virus NS3-4A heterocomplex serine protease activity expressed in human cells. Biochemistry 2005, 44, 6586–6596. [Google Scholar] [CrossRef] [PubMed]
- Phalaphol, A.; Thueng-in, K.; Thanongsaksrikul, J.; Poungpair, O.; Bangphoomi, K.; Sookrung, N.; Srimanote, P.; Chaicumpa, W. Humanized-VH/VHH that inhibit HCV replication by interfering with the virus helicase activity. J. Virol. Methods 2013, 194, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Thueng-in, K.; Thanongsaksrikul, J.; Srimanote, P.; Bangphoomi, K.; Poungpair, O.; Maneewatch, S.; Choowongkomon, K.; Chaicumpa, W. Cell penetrable humanized-VH/VHH that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLOS ONE 2012, 7, e49254. [Google Scholar] [CrossRef] [PubMed]
- Poungpair, O.; Pootong, A.; Maneewatch, S.; Srimanote, P.; Tongtawe, P.; Songserm, T.; Tapchaisri, P.; Chaicumpa, W. A human single chain transbody specific to matrix protein (M1) interferes with the replication of influenza A virus. Bioconjug. Chem. 2010, 21, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Thueng-in, K.; Thanongsaksrikul, J.; Jittapisutthikul, S.; Seesuay, W.; Chulanetra, M.; Sakolvaree, Y.; Srimanote, P.; Chaicumpa, W. Interference of HCV replication by cell penetrable human monoclonal scFv specific to NS5B polymerase. mAbs 2014, 6, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Yodsheewan, R.; Maneewatch, S.; Srimanote, P.; Thueng-in, K.; Songserm, T.; Dong-Din-On, F.; Bangphoomi, K.; Sookrung, N.; Choowongkomon, K.; Chaicumpa, W. Human monoclonal ScFv specific to NS1 protein inhibits replication of influenza viruses across types and subtypes. Antiviral Res. 2013, 100, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Qiao, Y.; Ran, Z.; Wang, T. Up-regulation and pre-activation of TRAF3 and TRAF5 in inflammatory bowel disease. Int. J. Med. Sci. 2013, 10, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Pieper, U.; Webb, B.M.; Barkan, D.T.; Schneidman-Duhovny, D.; Schlessinger, A.; Braberg, H.; Yang, Z.; Meng, E.C.; Pettersen, E.F.; Huang, C.C.; et al. ModBase, a database on annotated comparative protein structure models and associated resources. Nucleic Acid Res. 2011, 39, D465–D474. [Google Scholar]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acid Res. 2003, 31, 3316–3319. [Google Scholar]
- I-Tasser Online, Protein Structure & Function Prediction—Google Search. Available online: http://zhanglab.ccmb.med.umic.edu/I-TASSER/ (accessed on 1 March 2014).
- ModRefiner: High-resolution Protein Structure Refinement and Relaxation by Energy Minimization. Available online: http://zhanglab.ccmb.med.umich.edu/ModRefiner/ (accessed on 3 March 2014).
- FG-MD: High-Resolution Proteins Structure Refinement by Fragment-Guided MD Simulation. Available online: http://zhanglab.ccmb.med.umich.edu/FG-MD/ (accessed on 3 March 2014).
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK webserver for data-driven biomolecular docking. Nat. Protocols 2010, 5, 883–897. [Google Scholar]
- Chavanayarn, C.; Thanongsaksrikul, J.; Thueng-in, K.; Bangphoomi, K.; Sookrung, N.; Chaicumpa, W. Humanized-single domain antibodies (VH/VHH) that bound specifically to Naja kaouthia phospholipase A2 and neutralized the enzymatic activity. Toxins 2012, 4, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, A.; Decanniere, K.; Muyldermans, S.; Wyns, L. Antigen specificity and high affinity binding provided by one single loop of a camel single-domain antibody. J. Biol. Chem. 2001, 276, 26285–26290. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yamaji, K.; Masuho, Y.; Yokota, T.; Inoue, H.; Sudo, K.; Satoh, S.; Shimotohno, K. Identification of the sequence on NS4A required for enhanced cleavage of the NS5A/5B site by hepatitis C virus NS3 protease. J. Virol. 1996, 70, 127–132. [Google Scholar] [PubMed]
- Kim, J.; Morgenstern, K.; Lin, C.; Fox, T.; Dwyer, M.; Landro, J.; Chambers, S.; Markland, W.; Lepre, C.; O’Malley, E.; Harbeson, S.; Rice, C.; Murcko, M.; Caron, P.; Thomson, J. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Jacobsen, H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol. 1993, 67, 3835–3844. [Google Scholar] [PubMed]
- Tomei, L.; Failla, C.; Santolini, E.; de Francesco, R.; La Monica, N. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J. Virol. 1993, 67, 4017–4026. [Google Scholar] [PubMed]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Wilkinson, T.; Koch, J.O. Complex formation between the NS3 serine-type proteinase of the hepatitis C virus and NS4A and its importance for polyprotein maturation. J. Virol. 1995, 69, 7519–7528. [Google Scholar] [PubMed]
- Bartenschlager, R.; Ahlborn-Laake, L.; Yasargil, K.; Mous, J.; Jacobsen, H. Substrate determinants for cleavage in cis and in trans by the hepatitis C virus NS3 proteinase. J. Virol. 1995, 69, 198–205. [Google Scholar] [PubMed]
- Cento, V.; Mirabelli, C.; Salpini, R.; Dimonte, S.; Artese, A.; Costa, G.; Mercurio, F.; Svicher, V.; Parrotta, L.; Bertoli, A.; et al. HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLOS ONE 2012, 7, e39652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J. Learning from the messengers: Innate sensing of viruses and cytokine regulation of immunity—Clues for treatments and vaccines. Viruses 2013, 5, 470–527. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.M.; Ikeda, M.; Ray, S.C.; Gale, M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R.; Ikeda, M.; Lemon, S.M.; Gale, M. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Huang, B.; Lu, J.; Liu, Y.J.; Zhong, J. Hepatitis C virus NS3/4A protease blocks IL-28 production. Eur. J. Immunol. 2012, 42, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jittavisutthikul, S.; Thanongsaksrikul, J.; Thueng-in, K.; Chulanetra, M.; Srimanote, P.; Seesuay, W.; Malik, A.A.; Chaicumpa, W. Humanized-VHH Transbodies that Inhibit HCV Protease and Replication. Viruses 2015, 7, 2030-2056. https://doi.org/10.3390/v7042030
Jittavisutthikul S, Thanongsaksrikul J, Thueng-in K, Chulanetra M, Srimanote P, Seesuay W, Malik AA, Chaicumpa W. Humanized-VHH Transbodies that Inhibit HCV Protease and Replication. Viruses. 2015; 7(4):2030-2056. https://doi.org/10.3390/v7042030
Chicago/Turabian StyleJittavisutthikul, Surasak, Jeeraphong Thanongsaksrikul, Kanyarat Thueng-in, Monrat Chulanetra, Potjanee Srimanote, Watee Seesuay, Aijaz Ahmad Malik, and Wanpen Chaicumpa. 2015. "Humanized-VHH Transbodies that Inhibit HCV Protease and Replication" Viruses 7, no. 4: 2030-2056. https://doi.org/10.3390/v7042030
APA StyleJittavisutthikul, S., Thanongsaksrikul, J., Thueng-in, K., Chulanetra, M., Srimanote, P., Seesuay, W., Malik, A. A., & Chaicumpa, W. (2015). Humanized-VHH Transbodies that Inhibit HCV Protease and Replication. Viruses, 7(4), 2030-2056. https://doi.org/10.3390/v7042030